GENEB:为何基因组模型难以比较 GENEB: Why Genomic Models Are Hard to Compare
提出GENEB基准评估40个基因组模型在100任务上的表现,揭示聚合排名的不稳定性
前置知识
Probing Protocol
探针协议是一种模型评估方法,通过冻结预训练模型的参数,仅训练轻量级分类器(如逻辑回归)来测试模型表征质量。这种方法可以隔离表征学习能力,避免任务特定微调的混淆因素,从而实现跨架构和预训练方案的公平比较。GENEB使用逻辑回归(最大迭代1000)在1-shot、10-shot和全数据 regime 下进行探针评估。
GENEB的核心评估方法,理解它对于理解实验设计和结果解读至关重要。
Matthews Correlation Coefficient (MCC)
Matthews相关系数是一个用于评估二分类性能的指标,定义为MCC = (TP * TN - FP * FN) / sqrt((TP+FP)(TP+FN)(TN+FP)(TN+FN)),其中TP、TN、FP、FN分别表示真阳性、真阴性、假阳性和假阴性。MCC范围在[-1, 1]之间,1表示完美预测,0表示随机猜测,-1表示完全相反预测。MCC对类别不平衡具有鲁棒性,是基因组评估的标准指标。
GENEB的主要评估指标,理解它的计算和特性对于解读实验结果和模型性能比较非常重要。
Foundation Model
基础模型是在大规模数据上预训练的神经网络,能够通过下游任务微调或探针评估适应多种应用。在基因组学中,这些模型通常在DNA序列上训练,学习生物学相关的表征,如DNA-GPT(生成式)、GENOMEOCEAN(多物种)、EVO(进化建模)等。它们在架构上包括Transformer编码器、解码器、状态空间模型(如Mamba)和混合架构。
GENEB评估的对象,理解基础模型的概念和不同架构的特点对于理解论文动机和实验设计很重要。
研究动机
基因组机器学习领域在过去十年迅速扩张,产生了大量异构的模型、架构和训练范式,但这种扩张没有伴随着相应的方法论基础设施来支持比较。Figure 1通过有向图展示了当前领域状态:模型在分散的基准上评估,在不兼容的协议下比较,经常在狭窄定义的设置中报告为最先进技术。这导致很难回答基本问题,例如DNA-GPT、GENOMEOCEAN和EVO等广泛讨论的模型之间如何直接比较。同一个模型在一个背景下被描述为重大突破,在另一个背景下则表现不佳,这反映的不是矛盾证据,而是缺乏共同的评估框架。随着模型规模和可见性快速增长,关于优越性和泛化能力的主张变得更加大胆,但裁决这些主张的方法论基础却没有跟上步伐,导致模型能力的主张与可通过可重现跨模型评估建立的内容之间存在差距。
本文的目标是GENEB的目标是提供一个大规模、受控的基准,对40个基因组基础模型在跨越13个功能类别的100个DNA分类任务上进行统一评估。通过在完整的任务套件上以统一的基于探针的协议评估所有模型,GENEB实现跨架构、分词和预训练数据的匹配比较,并产生完整的性能矩阵,揭示任务相关的权衡。GENEB计划作为公共基准发布,评估结果托管在Hugging Face上,作为社区参考点,类似于NLP中的MTEB。这使得结果在模型和任务之间直接可比,为评估基因组机器学习的进展建立了共享参考点。
与已有工作不同的是,GENEB的独特切入角度是系统性地识别和量化当前基因组模型比较实践中的根本问题。与通常评估少数代表性架构或提供动态但基线有限和不断演变的排行榜的先前比较基准研究(如OmniGenBench)不同,GENEB提供了前所未有的规模和受控性。它不仅报告聚合性能,还明确暴露任务级别的权衡,并展示聚合排行榜的不稳定性:模型排名在任务类别间急剧变化,规模仅提供适度且不一致的增益,架构和预训练对齐经常超过参数计数。更重要的是,GENEB通过受控对匹配配对(匹配架构、分词和规模)的分析,隔离了预训练语料库效应,这是先前工作没有系统做到的。这使得GENEB能够得出关于设计模式(如多物种预训练、Transformer与SSM架构、分词策略)对性能影响的实践建议,而不仅仅是一个排行榜。
核心方法
GENEB采用基于嵌入的探针协议评估基因组基础模型,冻结序列表征并使用轻量级分类器进行评估,隔离表征质量并实现跨架构和训练方案的受控比较。评估覆盖跨越多个功能类别的多样化基因组预测任务,包括转录因子结合、增强子、启动子、剪接位点、染色质可及性、组蛋白修饰、lncRNA、DNA甲基化、小鼠增强子、病毒/噬菌体、物种分类、调控和编码/非编码区分。对于每个任务,冻结嵌入被用作逻辑回归(最大迭代1000)的特征,在1-shot、10-shot和全数据regime下评估,结果在五个固定随机种子{13, 17, 42, 123, 997}上平均。报告Matthews相关系数(MCC),这是基因组评估的标准指标,对类别不平衡具有鲁棒性。超过10的5次方个序列的任务被子采样,基于使用GENOMEOCEAN嵌入的实证分析显示MCC在此大小后稳定化,因此将10的5次方作为实用上限。
GENEB的核心创新是提供一个统一、受控的比较框架,揭示聚合排行榜的不稳定性并隔离不同设计选择(规模、架构、分词、预训练数据)的贡献。与先前评估小模型子集或使用异构协议的研究不同,GENEB在相同任务集和评估协议下评估40个模型,使得结果直接可比。更关键的是,GENEB通过受控比较隔离特定设计选择的效应:通过匹配预训练语料库和分词来比较架构(如Transformer编码器与Mamba),通过匹配架构和分词来比较预训练数据类型(如人类与多物种),通过匹配架构和预训练数据来比较分词方案(如BPE与k-mer)。这种受控方法使得GENEB能够识别模式,如Transformer在匹配条件下优于状态空间模型、多物种预训练在真核任务上优于人类特异性预训练、以及分词效果与模型家族交互而非全局排序。这些发现基于完整性能矩阵和排名的统计分析(如Spearman相关系数),而不仅仅是聚合平均。
方法步骤详情
GENEB的评估流程包括四个主要步骤。首先,对于每个模型,在预训练期间冻结权重,对每个任务的输入DNA序列提取嵌入表示。嵌入提取使用预训练模型的默认配置,包括分词方案和上下文窗口。其次,对于每个任务,使用冻结嵌入训练逻辑回归分类器,最大迭代次数为1000,使用默认正则化。在few-shot regimes下,从完整训练集中随机抽取指定数量的样本(1-shot或10-shot)。第三,在测试集上评估训练好的分类器,计算Matthews相关系数(MCC)。对于所有任务,测试集是固定且不变的,确保跨模型比较的公平性。最后,对于每个模型、任务和regime(1-shot、10-shot、全数据),在五个固定随机种子{13, 17, 42, 123, 997}上重复训练和评估,并报告平均MCC。对于涉及多个任务的类别,计算macro-averaged MCC(所有任务的平均)和micro-averaged MCC(所有实例的平均),并在分析中主要使用macro-averaged MCC以避免大类别(如组蛋白修饰30个任务、启动子22个任务)主导结果。结果被组织成完整的性能矩阵(40个模型乘以100个任务乘以3个regimes),用于后续分析和可视化。
技术新颖性
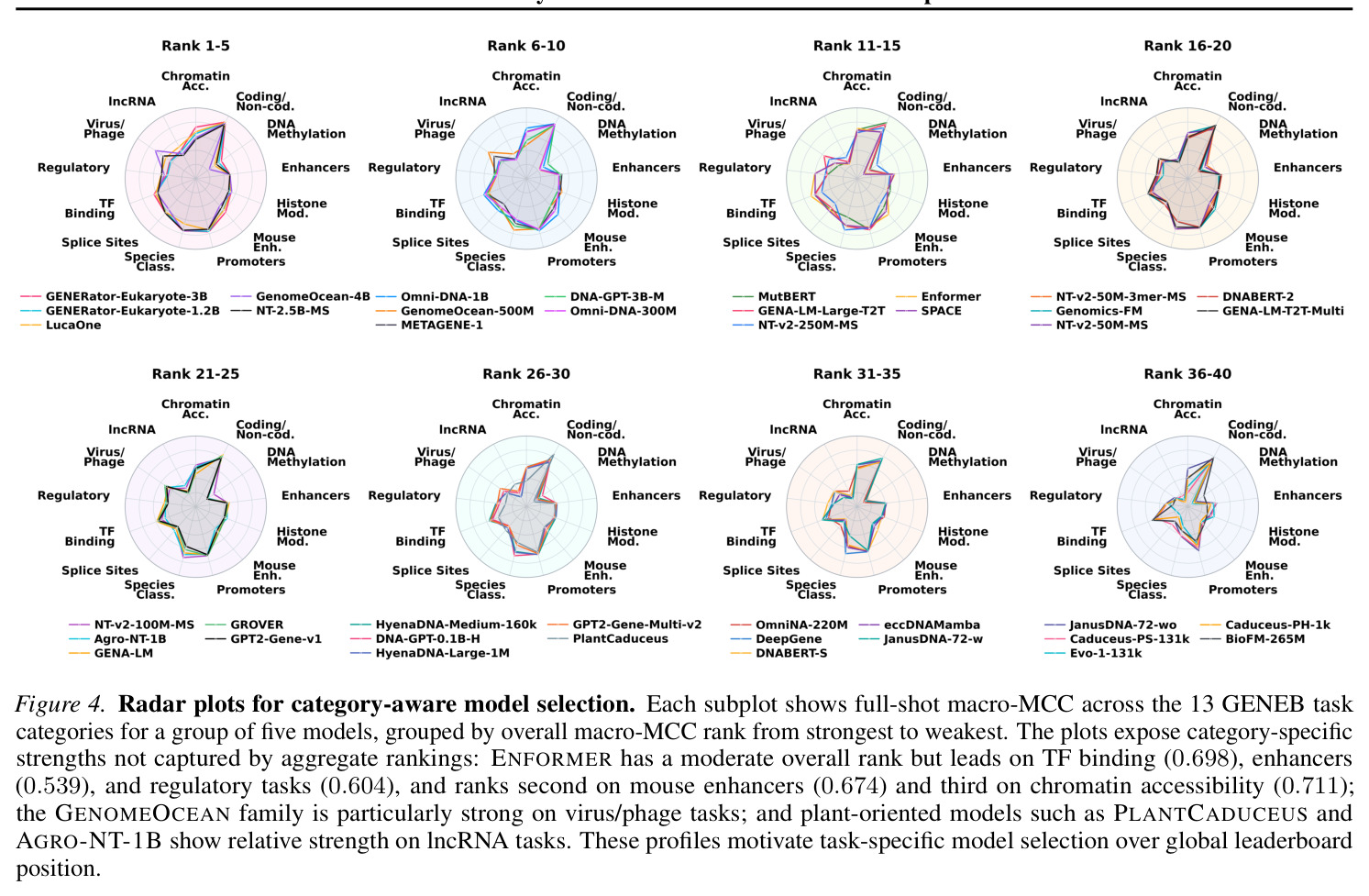
GENEB的技术新颖性体现在多个方面。首先,它是迄今为止最大规模的基因组基础模型基准,评估40个模型在100个任务上的表现,比先前工作大一个数量级。其次,GENEB是第一个系统性地使用受控比较来隔离设计选择效应的基因组基准,通过匹配配对(匹配架构、分词和规模)分析预训练语料库效应,这在基因组机器学习中是独特的。第三,GENEB引入了详细的few-shot评估(1-shot和10-shot),并揭示了一个反向性能模式:在full-shot上表现弱的模型在few-shot下的绝对下降小,不是因为它们更稳健,而是因为它们的full-shot上限已经很低。第四,GENEB通过实证分析(如在附录E.1中使用MLP探针验证线性探针下模型排名的稳定性)和限制讨论(如冻结表征可能低估特定任务微调的可达性能、分词与池化交互未完全解缠)展示了方法论的严谨性。最后,GENEB不仅报告聚合性能,还提供了实践建议,针对不同任务类别和计算约束推荐特定模型(如紧凑部署使用MUTBERT、表观基因组谱任务使用ENFORMER和SPACE),这使得GENEB对实践者有直接价值。
实验结果
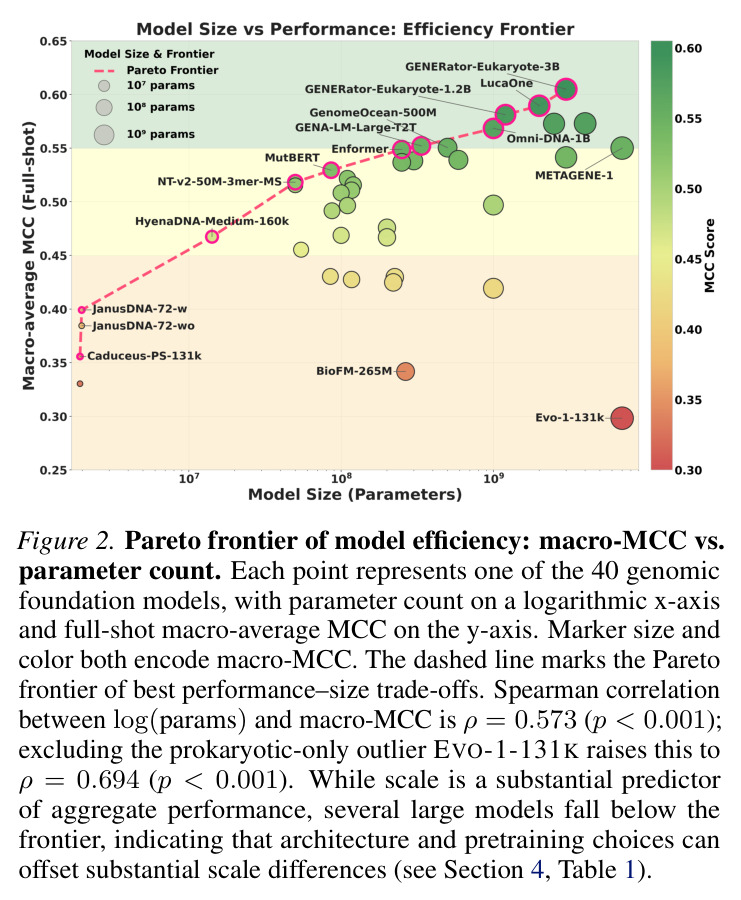
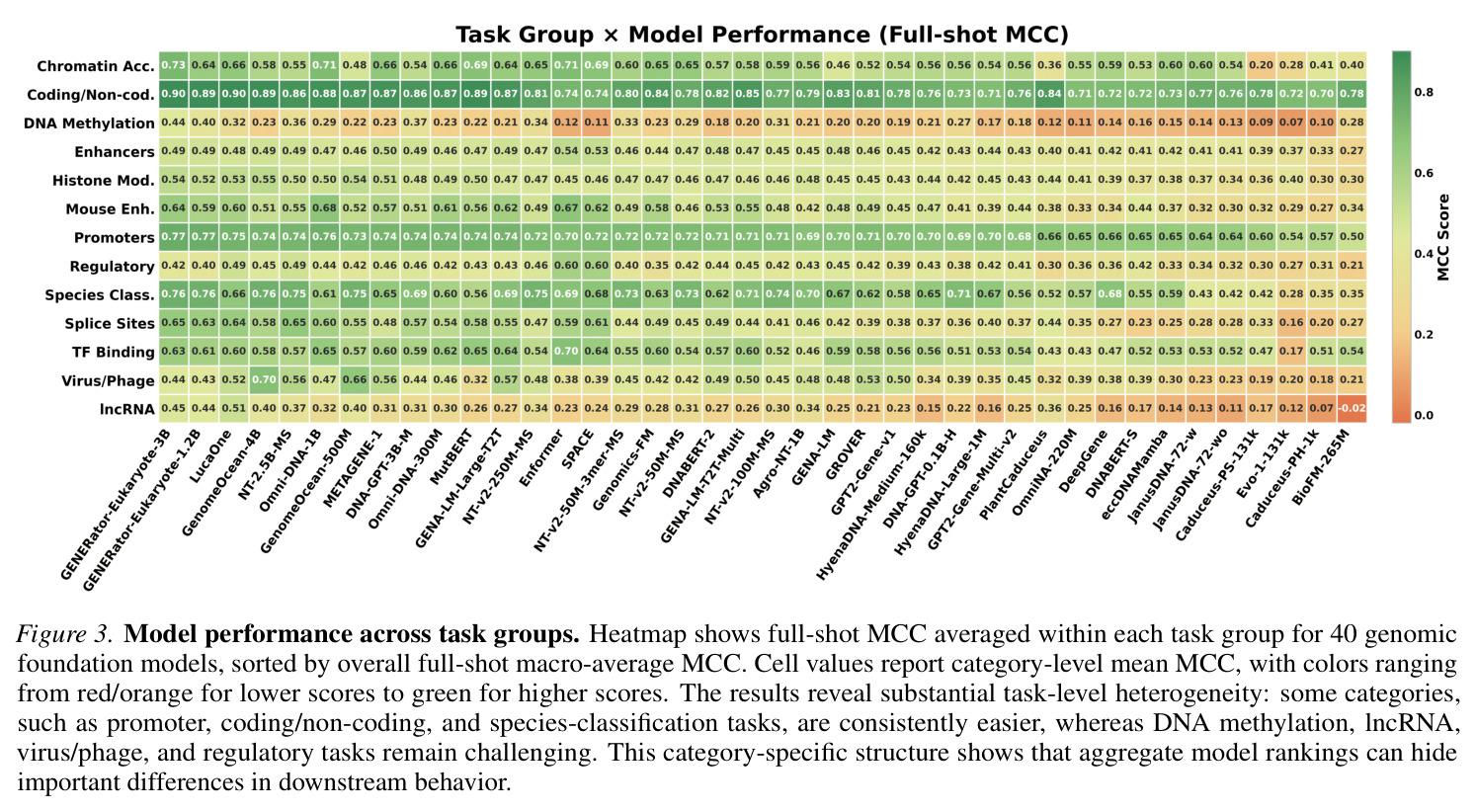
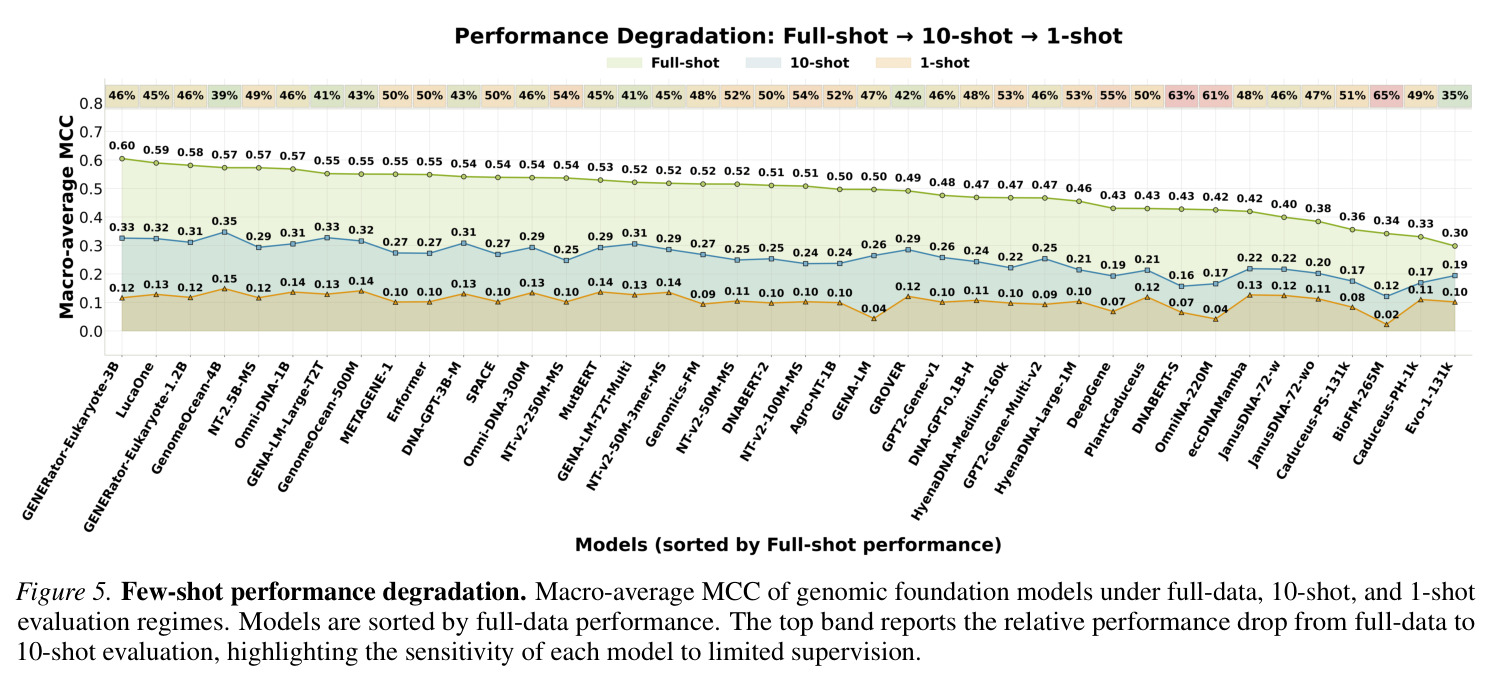
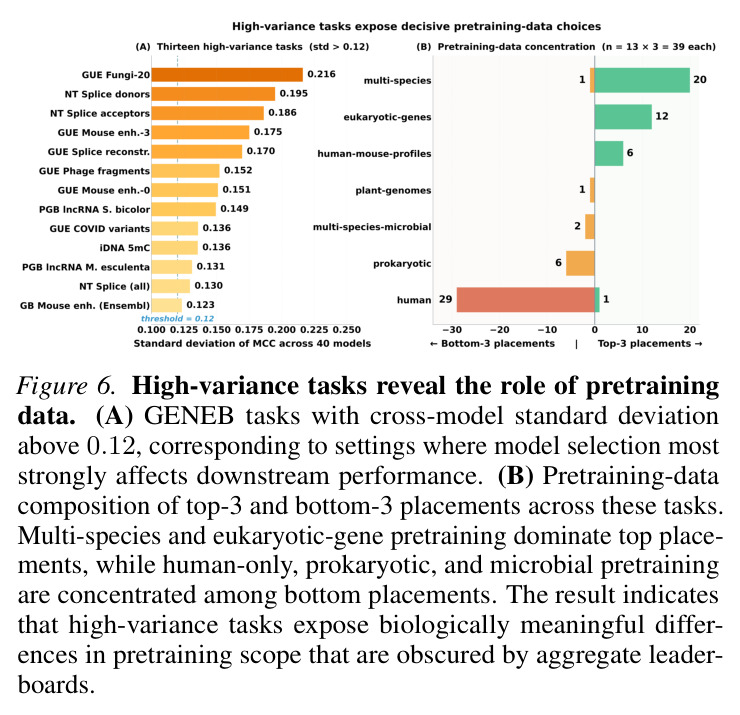
GENEB的系统性分析揭示了关于基因组模型比较的几个关键发现。首先,模型规模与聚合性能之间存在统计显著且实质性的关联(Spearman相关系数等于0.573,p值小于0.001,Figure 2),在排除仅原核生物的离群值EVO-1-131K后增强到0.694(p值小于0.001)。然而,规模不是完美的类别级结果预测器:在36个领域内模型中,识别出25个实例,其中至少小5倍的模型在聚合MCC上超过更大的对应模型。代表性示例是MUTBERT(86M)超过ECCDNAMAMBA(537M)0.110 macro-MCC,尽管有6.2倍的规模差异。其次,架构在控制条件下有实质影响:Transformer模型通常优于评估的状态空间替代方案。具体来说,GENOMEOCEAN-500M(Transformer-decoder)超过ECCDNAMAMBA(Mamba)0.131 macro-MCC(0.550 vs 0.419),OMNI-DNA-1B超过ECCDNAMAMBA 0.149(0.568 vs 0.419)。在Transformers内部,编码器模型在所有六个匹配配对中超过解码器(Appendix E.3),在匹配的多物种/BPE条件下GENA-LM-LARGE-T2T超过OMNINA-220M的最大差距为0.127 MCC(0.552 vs 0.425)。第三,架构依赖的间隙在需要跨物种泛化的任务上特别明显(Figure 3和4)。在病毒/噬菌体上,GENOMEOCEAN-500M(Transformer-decoder)超过ECCDNAMAMBA(Mamba)0.355 macro-MCC(0.657 vs 0.302)。在小鼠增强子上,OMNI-DNA-1B超过ECCDNAMAMBA 0.305(0.675 vs 0.370),GENA-LM-LARGE-T2T在匹配条件下超过OMNINA-220M 0.284。这些类别级间隙比聚合参数层增益(超过1B和低于200M参数之间的模型0.075 macro-MCC)大几倍。第四,few-shot稳健性揭示了反向性能模式。跨所有40个模型,平均macro-MCC从full-shot的0.488下降到10-shot的0.253和1-shot的0.106(Figure 5),对应相对减少48.2%和78.2%。每模型相对10-shot下降从35%(EVO-1-131K)到65%(BIOFM-265M)不等(中位数48%)。五个最小的绝对下降发生在EVO-1-131K(下降等于0.196)、CADUCEUS-PH-1K(0.220)、JANUSDNA-72-WO(0.272)、CADUCEUS-PS-131K(0.272)和JANUSDNA-72-W(0.275)——都是full-shot中最弱的模型之一。顶级full-shot表现者显示相反模式:GENERATOR-EUKARYOTE-3B(下降等于0.489)、GENERATOR-EUKARYOTE-1.2B(0.463)、LUCAONE(0.461)和NT-2.5B-MS(0.456)都超过0.42的绝对下降。这种模式不表示弱模型更稳健:小的绝对下降反映低的full-shot上限,留给进一步下降的空间有限。EVO-1-131K说明性——其低上限源于域不匹配,而不是few-shot regime本身。第五,GENEB的困难前沿显示28个100任务的平均MCC低于0.35(Figure 3),由4mC甲基化预测(G. subterraneus 0.061;E. coli 0.103;G. pickeringii 0.107)和植物lncRNA识别(S. lycopersicum 0.221;G. max 0.228;T. aestivum 0.238)主导。即使这些类别上最强的模型,GENERATOR-EUKARYOTE-3B在4mC任务上仅达到0.206-0.477,LUCAONE在植物lncRNA上达到0.417-0.629。类别级缩放分析显示这种困难性不是纯参数计数问题:13个类别中11个显示在p值小于0.05下正缩放显著,但强度变化很大(相关系数从DNA甲基化的0.346到组蛋白修饰的0.587)。两个类别显示无显著缩放:物种分类(相关系数等于0.308,p值等于0.054)和染色质可及性(相关系数等于0.240,p值等于0.136)。即使缩放统计显著,GENEB的困难前沿显示仅缩放不能关闭绝对性能差距;这些任务上的进展将需要预训练语料库设计、归纳偏置或任务特定监督的互补进展。

查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| Transcription Factor Binding | macro-MCC (full-shot) | ENFORMER 0.698 | OMNI-DNA-1B 0.647 | +0.051 (7.9%) |
| Virus/Phage Classification | macro-MCC (full-shot) | GENOMEOCEAN-4B 0.697 | ECCDNAMAMBA 0.302 | +0.395 (131%) |
| Species Classification | macro-MCC (full-shot) | GENOMEOCEAN-4B 0.762 | NT-V2-100M-MS 0.741 | +0.021 (2.8%) |
| DNA Methylation | macro-MCC (full-shot) | GENERATOR-EUKARYOTE-3B 0.440 | DNA-GPT-3B-M 0.367 | +0.073 (20%) |
| Chromatin Accessibility (SSM advantage) | macro-MCC (full-shot) | ECCDNAMAMBA 0.599 | GENOMEOCEAN-500M 0.475 | +0.124 (26%) |
局限与改进
作者承认了GENEB的几个局限性。首先,GENEB对需要显式建模非常长程调控相互作用(大于10 kb)的任务代表性不足。结果,具有显式长上下文能力的模型(HYENADNA-LARGE-1M、CADUCEUS-PS-131K、EVO-1-131K)在其架构先验最可能产生差异化增益的regime下没有得到充分测试。Appendix D.3中提供了考虑但排除的长程数据集和模型上下文长度约束的详细枚举。其次,任务选择受可用数据集和现有基准的约束。一些构成任务可能是嘈杂或弱定义的,特别是在GENEB中识别的困难前沿(DNA甲基化、植物lncRNA),其中标签质量和监督信号跨来源变化。随着基因组基准的成熟,需要进一步的任务细化和改进。第三,并非所有基因组基础模型都能由于权重不可用、不兼容管道或计算约束而被包括在内。排除模型和纳入标准在Appendix D中讨论。第四,GENEB的13个类别中只有病毒/噬菌体分类反映非真核域;原核基因预测、微生物基因组组装验证和CRISPR系统表征当前未表示。因此,聚合GENEB排名是原生或病毒基因组学中性能的不可靠代理(参见Section 4的域不匹配段落)。最后,GENEB使用线性探针评估冻结表征,这实现了40模型集上嵌入质量的受控比较,但可能低估可通过任务特定微调实现的性能。附录E.1中的实证分析显示线性探针下的模型排名与非线性MLP探针高度一致;但这种稳定性是否扩展到完整任务特定微调仍是一个开放问题。此外,单核苷酸和k-mer分词为固定输入窗口产生比BPE长得多的标记序列,因此池化选择(平均、注意力加权或最终标记)可能有利于不同方案。GENEB全程使用平均池化;池化-分词交互未完全解缠。
独立分析的弱点
从独立分析角度,GENEB有几个可以改进的弱点。首先,GENEB的任务套件严重偏向真核基因现象(13个类别中12个),为预训练在原核或微生物语料库上的模型创造了结构性劣势。虽然作者承认这一点,但它限制了GENEB在原生或病毒基因组学中的实用性。改进方向是扩展任务套件以包括更多原生和病毒任务,如原核基因预测、CRISPR系统表征和噬菌体宿主预测。其次,GENEB使用冻结表征和线性探针,这可能低估了通过任务特定微调可实现的性能,特别是对于具有不同归纳偏置的架构(如Mamba的序列建模与Transformer的全局注意力)。改进方向是补充冻结探针评估与有限微调评估(例如冻结预训练权重但训练任务特定头部)或完整端到端微调,以评估微调的增益。第三,GENEB的任务选择受可用数据集和现有基准的约束,一些任务可能是嘈杂或弱定义的,特别是在困难前沿(DNA甲基化、植物lncRNA)。改进方向是系统性地评估和细化任务质量,通过交叉验证多个数据源、手动注释检查和与领域专家合作。第四,GENEB的few-shot评估使用简单随机抽样,这可能不能反映真实世界的few-shot场景,其中样本可能是主动选择或以特定方式标记。改进方向是探索few-shot策略,如主动学习、半监督学习和跨任务迁移。最后,GENEB的评估主要基于MCC,这是一个二分类指标。对于多类任务或回归任务(如表达水平预测),MCC可能不是最佳指标。改进方向是扩展指标套件以包括领域特定指标,如AUPRC(用于不平衡分类)、F1-score(用于精度/召回权衡)和RMSE(用于回归任务)。
未来方向
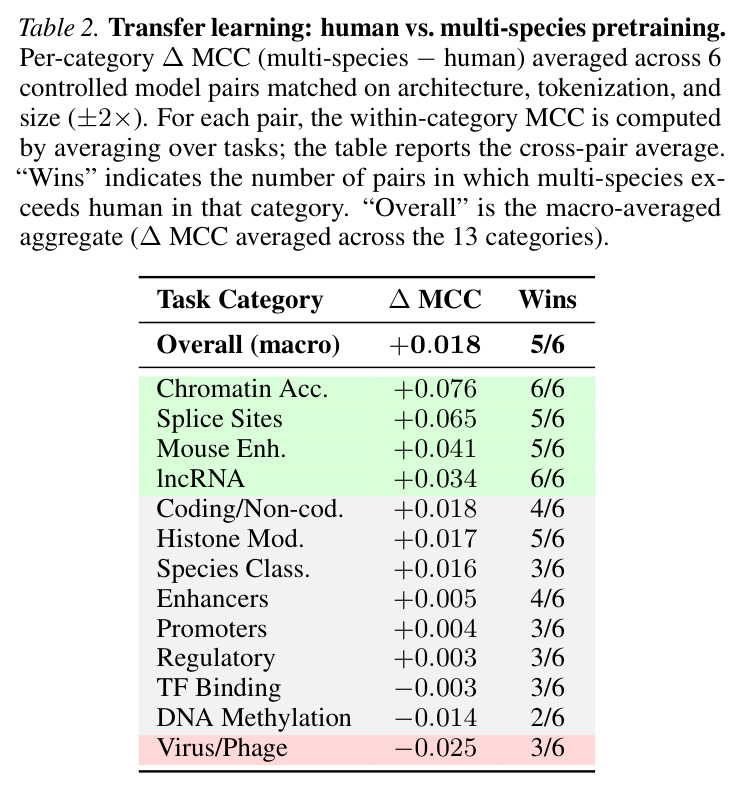
基于GENEB的发现,有几个有前景的未来研究方向。首先,GENEB的困难前沿(DNA甲基化、植物lncRNA)识别了需要预训练语料库设计、归纳偏置或任务特定监督互补进展的领域。具体方向包括设计针对甲基化模式的位置特定归纳偏置(因为甲基化信号是位置特定的)、构建更高质量的植物lncRNA数据集(因为当前任务平均MCC低于0.35)和探索任务特定架构(如CNN-Transformer混合用于长程调控)。其次,GENEB发现多物种预训练在真核任务上一致优于人类特异性预训练(Table 2),特别是在染色质可及性(增益等于0.076)、剪接位点(增益等于0.065)和小鼠增强子(增益等于0.041)。未来工作可以探索预训练语料库的细粒度设计,例如针对任务类型的特异性物种子集、进化距离加权(更近缘物种获得更高权重)和功能注释(如仅预训练在编码序列上用于编码/非编码区分)。第三,GENEB发现架构在受控条件下有实质影响,Transformer通常优于Mamba,但有域特定例外(染色质可及性)。未来工作可以探索混合架构,结合Transformer的全局建模和SSM的长程建模优势,以及针对特定任务类型的架构搜索(如用于调控任务的CNN-Transformer混合)。第四,GENEB发现分词效果与模型家族交互而非全局排序,没有单一的偏好方案。未来工作可以探索自适应分词,根据任务类型和输入序列长度调整分词策略,以及端到端学习分词(而不是使用预定义的k-mer或BPE)。最后,GENEB的few-shot评估揭示了反向性能模式,暴露了聚合few-shot排行榜的结构局限性。未来工作可以开发领域感知的few-shot评估协议,考虑任务可解性与模型质量的分离,以及few-shot设置中的calibrated指标(如绝对性能相对于随机,而不是相对于full-shot)。
复现评估
GENEB的复现性有几个方面的评估。首先,论文声明GENEB将作为公共基准发布,评估结果托管在Hugging Face上,这将使社区能够验证和扩展结果。然而,论文没有明确提供代码和数据存储库,这对于完全复现是必要的。理想情况下,作者应提供:1)评估管道代码,包括嵌入提取、探针训练和指标计算;2)任务定义和数据处理脚本,确保跨模型的一致性;3)模型权重加载代码和预训练模型链接(对于公开可用的模型);4)实验配置和随机种子,确保结果可重现。其次,论文提到评估40个模型在100个任务上,涉及大量计算。虽然论文没有报告具体的计算资源使用,但估计这需要大量GPU时间,特别是对于大型模型(如GENOMEOCEAN-4B、EVO-1-131K)。这可能会限制资源有限的研究者复现完整基准的能力。第三,论文使用五个固定随机种子{13, 17, 42, 123, 997},这是好的实践,但论文没有报告每个模型-任务-regime对的标准差或置信区间,这对于评估结果的统计显著性很重要。第四,论文提到一些模型由于权重不可用、不兼容管道或计算约束而被排除(Appendix D),但没有详细说明排除标准,这可能会影响结果的可解释性。最后,论文的附录包括额外的分析(如非线性探针验证、正则化强度敏感性分析、macro与micro平均比较),这是好的透明性实践。总体而言,GENEB的复现性是中等的,但可以通过提供代码、详细报告计算资源使用和更完整的随机种子分析来改进。
论文图表
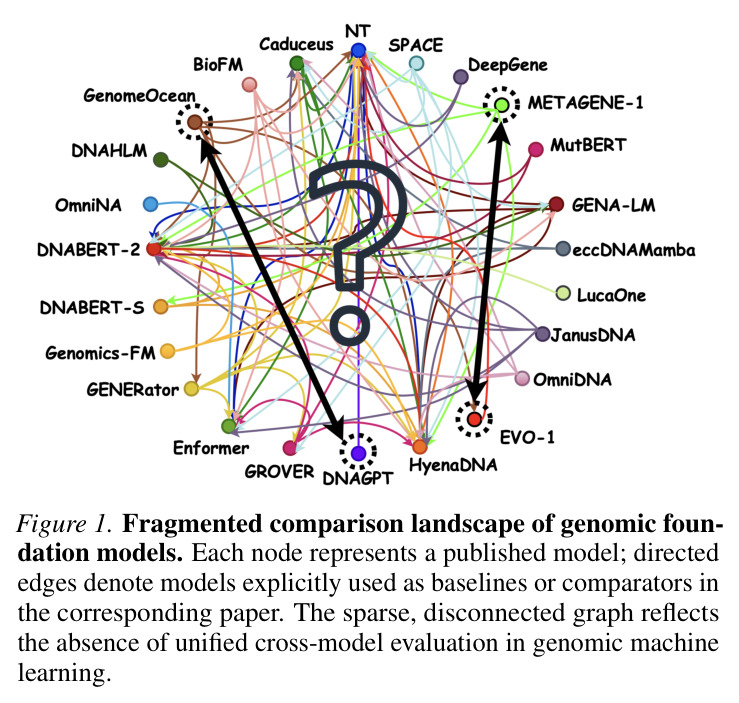
Figure 1通过有向图展示了基因组基础模型的分散比较格局。每个节点代表一个已发表的模型,有向边表示相应论文中明确用作基线或比较器的模型。稀疏、断开的图反映了基因组机器学习中缺乏统一的跨模型评估。图中的节点包括DNA-GPT、GENOMEOCEAN、EVO等广泛讨论的模型,但它们之间几乎没有直接连接,表明没有系统的比较研究将这些模型放在一起评估。
这张图对理解论文至关重要,因为它直观地展示了GENEB试图解决的核心问题:基因组模型比较的碎片化状态。它证明了当前评估实践的不系统性,不同论文在不同的基准和协议下评估模型,使得直接比较变得不可能。这为GENEB的动机提供了强有力的视觉支持。