单细胞CRISPR扰动的几何一致性揭示调控架构并预测细胞应激反应 Geometric coherence of single-cell CRISPR perturbations reveals regulatory architecture and predicts cellular stress
用方向一致性指标量化CRISPR扰动后细胞群体的几何稳定性。
前置知识
单细胞CRISPR扰动筛选(Perturb-seq)
Perturb-seq是结合CRISPR基因编辑与单细胞RNA测序的高通量实验范式。每个细胞既携带一个特定基因的扰动(敲除、过表达或抑制),又有完整的转录组读出,从而能在单细胞分辨率下观察同一扰动在不同细胞中的异质反应。典型数据集(如Replogle 2022)可在基因组规模上覆盖上千个基因扰动。
本文的核心数据集全部来源于Perturb-seq筛选,读者需要理解它的实验设计才能把握Sp指标为什么必须以单细胞层级而非群体均值来计算。
Waddington表观遗传景观
Waddington在1957年将细胞发育比喻为小球沿起伏表面的滚动:山谷代表稳定的细胞命运吸引子,脊背代表不稳定的中间态。景观中的曲率与势垒高度决定了细胞被扰动后是否能被通道化(canalized)回原状态。这是动力学系统拓扑的直观图像,近年被单细胞基因组学变得可直接测量。
本文的'几何税'框架完全建立在该景观隐喻之上:吸引子深度对应Sp高低,方向一致性对应细胞是否沿共同轨迹移动。没有这个直觉,论文的motivation会显得空泛。
PCA嵌入与余弦相似度
主成分分析(PCA)把高维表达矩阵线性投影到保留最大方差的少数坐标轴,本文用50维。给定两个向量,余弦相似度 $\cos\theta = \frac{d_i \cdot \bar{d}}{\|d_i\|\|\bar{d}\|}$ 衡量方向一致性,与向量长度无关,因此能单独捕捉'方向'信号。
Sp的公式本质上是PCA空间下的平均余弦相似度,理解这两个工具的线性假设与几何含义是看懂所有图表的门槛。
扰动响应分数(PS)
Song等人2025年提出的PS用scMAGeCK约束优化算法估计每个单细胞对扰动的响应强度,识别群体中'响应强烈'的细胞。它衡量的是个体细胞的响应幅度,不关心群体整体的方向组织。
PS是本文最重要的对照基线,Sp与PS的偏相关分析(控制效应量后)决定了两者是互补还是冗余。这是论文'差异性贡献'的核心。
未折叠蛋白反应(UPR)
UPR是内质网中错误折叠蛋白积累时启动的稳态应激通路,关键标志物包括HSPA5(BiP/GRP78)、DDIT3(CHOP)、XBP1、ATF4。当细胞被推入偏离正常吸引子的表达状态时,蛋白稳态失衡会触发UPR以恢复平衡。
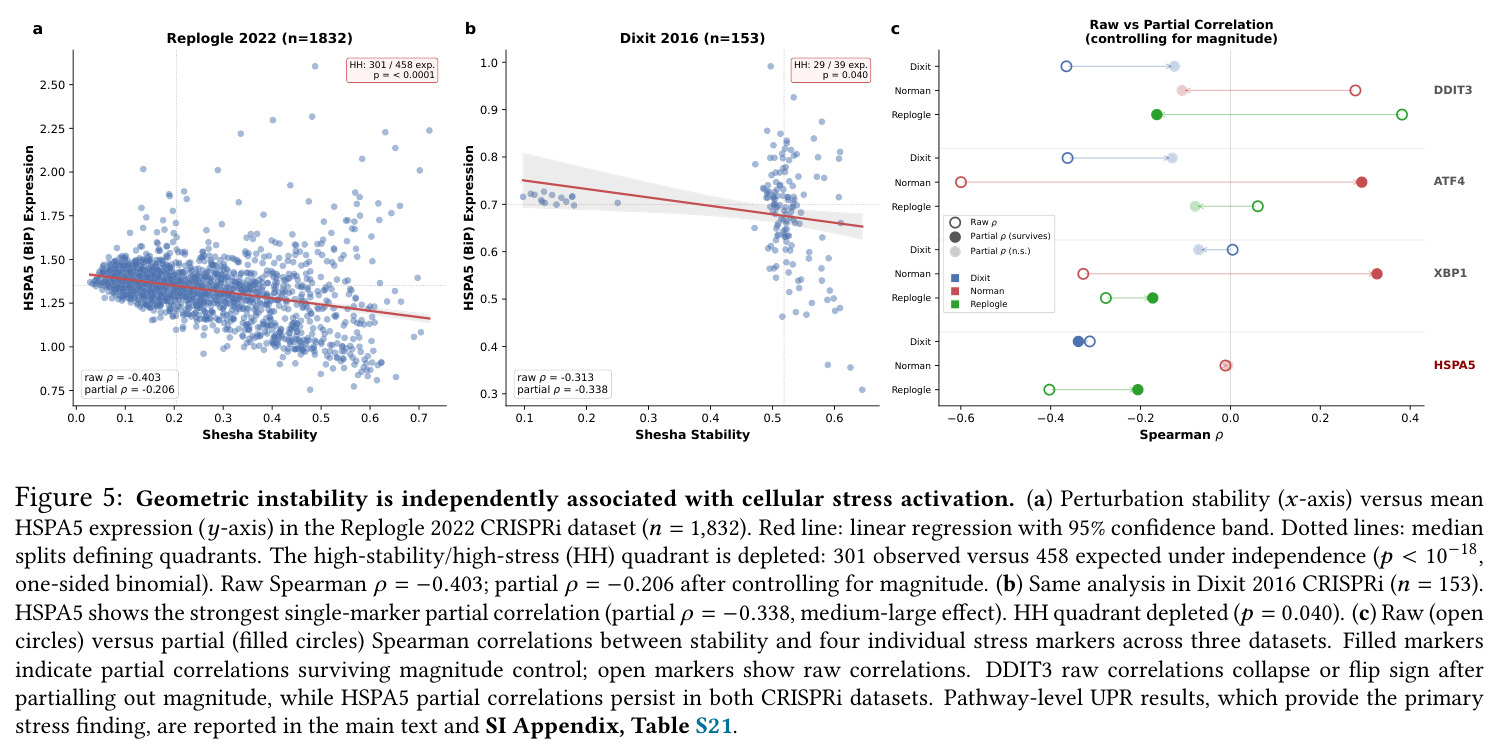
论文发现几何不稳定性与UPR通路激活在四个数据集上都呈一致的负偏相关,这是Sp从'描述性度量'升级为'功能性预测器'的关键证据。
研究动机
CRISPR-Cas9让我们能以序列级精度编辑基因组,但编辑成功的细胞仍可能漂移到非预期命运。当前评估框架如indel率、脱靶切割率、序列保真度只回答工程师的问题——'代码改对了吗?'——却不回答生物学家的问题——'改完之后的状态稳定吗?'。这种语法-语义鸿沟背后是三类失败:脱靶编辑、大型缺失/染色质碎裂、以及最根本的表型异质性。两项携带完全相同编辑的细胞可能一个分化、一个保持干性,差异源自它们在状态流形上的初始位置和局部几何。单细胞CRISPR筛选虽然能观察到这种几何信息,但标准分析只把它压缩成一个标量——效应量(控制与扰动后均值表达的欧氏距离),完全丢掉了'细胞们是否一起移动'这个维度。PCA/UMAP等线性降维进一步把高维流形压平,抹除了深谷与浅脊的曲率差异。两个群体在低维投影中可能看起来表型相似,实际却占据着能垒截然不同的位置。这是几何税概念的物理基础。
本文的目标是本文的具体目标有三层:第一,提出一个几何稳定性指标Shesha扰动稳定性 $S_p$,形式化为扰动细胞个体平移向量与群体平均方向之间的平均余弦相似度 $S_p = \frac{1}{n_p}\sum_{i=1}^{n_p} \frac{d_i \cdot \bar{d}}{\|d_i\|\|\bar{d}\|}$,其中 $d_i = x_i - \mu_{ctrl}$,让研究者能定量回答'细胞是否朝同一方向移动';第二,在5个数据集、2200+扰动中验证该指标与效应量的强相关性与在'几何税'极端情形下的解耦;第三,证明Sp在分裂半重复性、UPR应激通路预测、scGPT非线性嵌入三个独立场景中提供超越效应量和PS的增量信息。
与已有工作不同的是,已有方法从互补角度刻画扰动异质性但都未触及'方向一致性'。Nadig et al. 2025的TRADE通过转录组范围差异表达把扰动效应分解为共享与基因特异成分,但它工作在伪批量层面,关注的是基因级广度而非细胞级方向。Song et al. 2025的PS识别哪些个体细胞响应强烈,估计的是响应幅度而非群体几何组织。Replogle等大型数据集积累了大量单细胞分辨率数据却只用均值统计。本文抓住了被所有人忽略的角度——在'哪些细胞响应'之上的第二层问题:响应了的细胞是否走同一条路。这正是Waddington景观里'山谷深度'与'方向通道化'的几何对应物,没有这个度量就无法区分深谷中的KLF1型响应与浅脊上的CEBPA型响应。
核心方法
方法核心直觉非常简洁:想象一个房间里有一群人在音乐响起时各自迈步。如果所有人步伐方向一致(即使步幅有大有小),我们说这个群体'几何上稳定';如果他们朝四面八方散开,哪怕平均位移一样大,也是'几何不稳定'。把这个图像翻译到PCA空间:每个扰动细胞 $x_i$ 减去控制中心 $\mu_{ctrl}$ 得到平移向量 $d_i$,这些向量的方向一致性就是该扰动的'群体方向组织度'。技术路线上,作者分三步:先用标准scanpy流程把每个数据集预处理到50维PCA嵌入;然后为每个扰动计算所有细胞的平移向量及均值方向 $\bar{d}$,用公式 $S_p = \frac{1}{n_p}\sum_{i}\frac{d_i \cdot \bar{d}}{\|d_i\|\|\bar{d}\|}$ 得到0到1之间的稳定性分数(1=完全一致,0=完全发散);最后用LOESS(带宽分数0.3)而非线性回归计算效应量-稳定性残差,得到非线性'discordance'指标,识别那些'幅度大但方向散'的多效调控因子。该方法从一个通用的Shesha表征稳定性框架(Raju 2026a,b)借鉴了几何自一致性原理,但专门适配到扰动生物学语境。
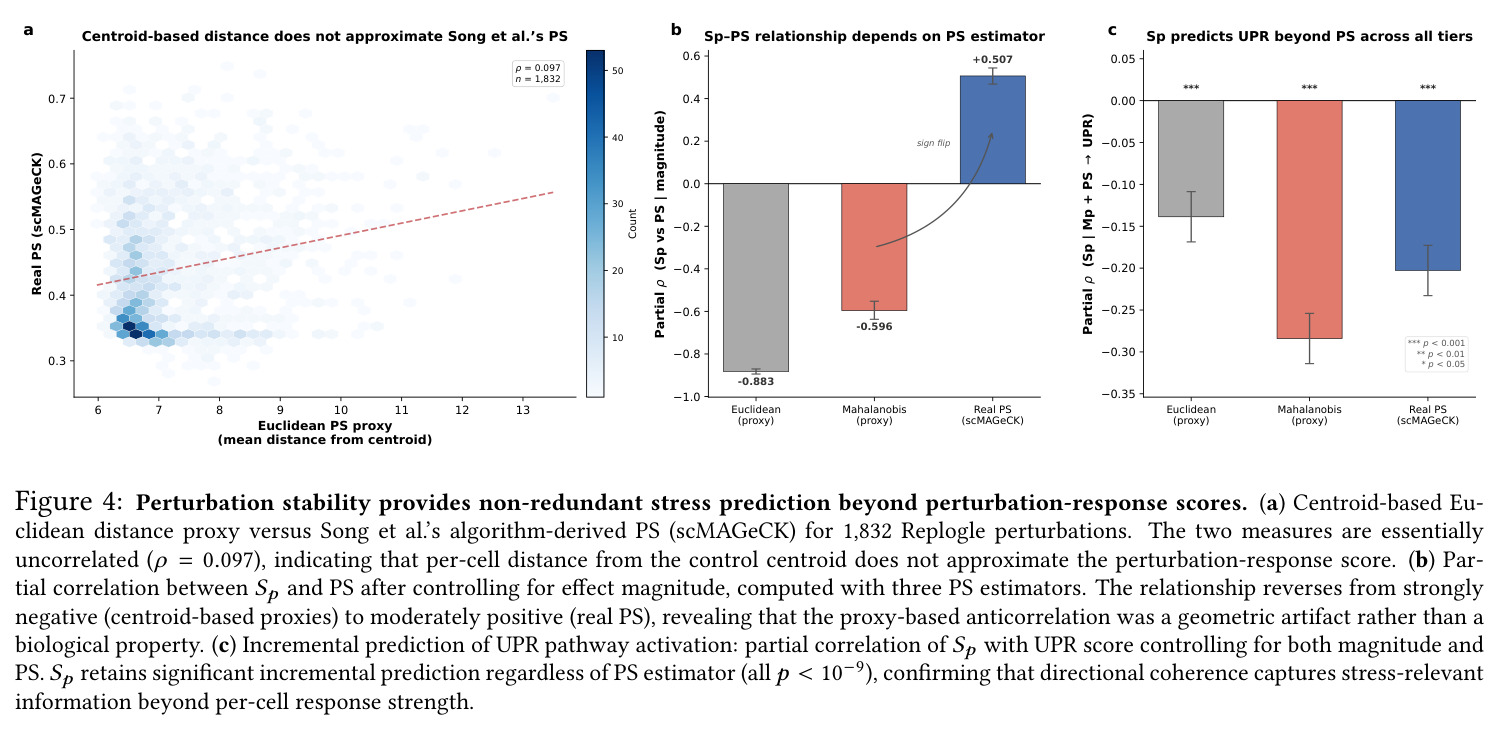
核心创新是把'群体是否一起移动'从直觉变成可计算的标量。本质区别于已有方法的关键点有三:其一,TRADE类方法在伪批量层面工作,把每个扰动压缩成基因级log-fold-change的分布,丢失细胞间方向信息;本文Sp操作在个体细胞平移向量上,保留几何结构的全部分辨率。其二,PS量化每个细胞响应的强度('谁响得猛'),Sp量化群体响应的方向组织('大家是否一起走'),二者问的是不同的问题;论文用三档PS对比揭示了基于质心的距离代理其实并不逼近真正的scMAGeCK PS($\rho=0.097$),之前看到的强负相关是几何重言式而非生物互补。其三,标准做法只用效应量(均值位移的欧氏范数)总结扰动,Sp通过余弦相似度天然剥离了幅度信息,专门捕捉方向——这是它能成为效应量'第二坐标轴'而非冗余指标的原因。
方法步骤详情
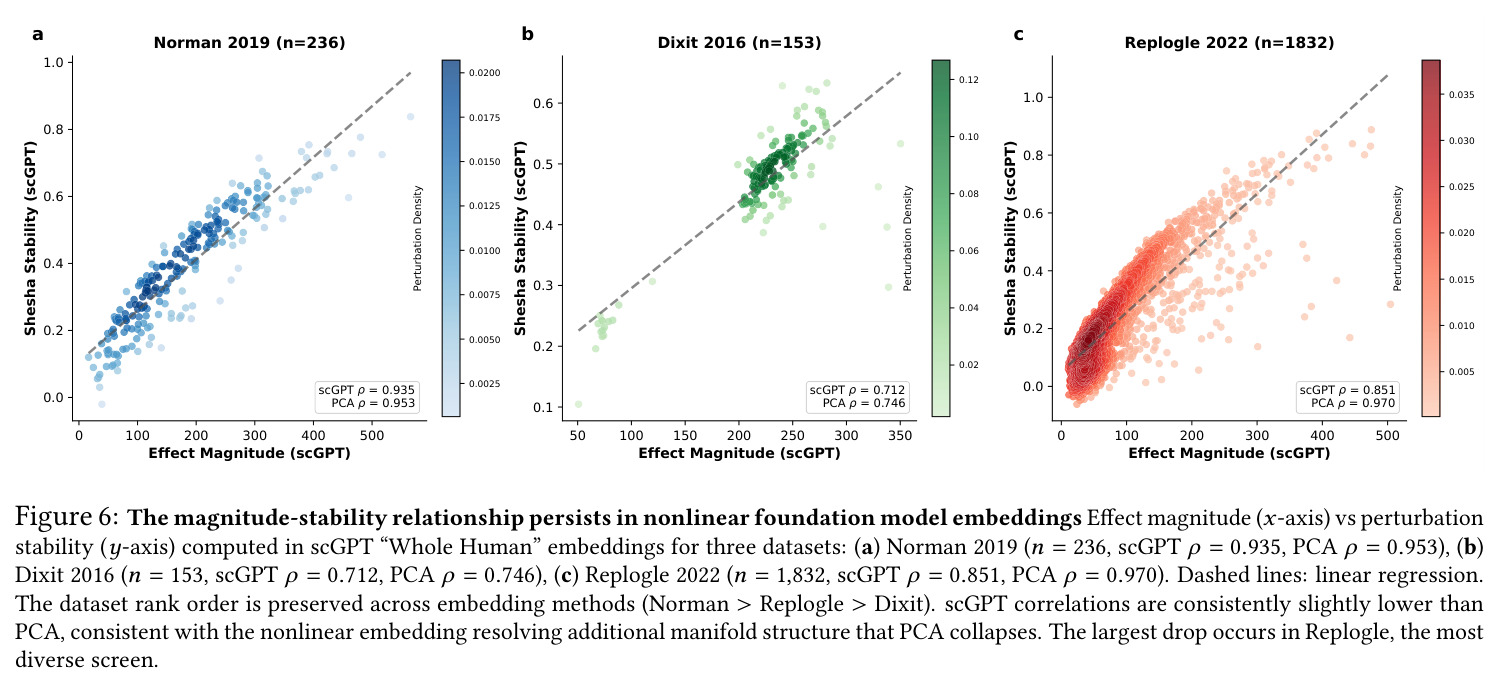
完整方法步骤如下:(1)数据获取与质控:从pertpy 1.0.4加载五个数据集(Norman 2019 CRISPRa K562 $n=236$;Adamson 2016 CRISPRi $n=8$;Dixit 2016 CRISPRi BMDC $n=153$;Papalexi 2021 pooled THP-1 $n=25$;Replogle 2022 CRISPRi K562 $n=1832$),过滤基因检出数<100的细胞,分别用scanpy.pp.normalize_total(Replogle/Adamson目标1e4)+log1p标准化,选前2000高变基因,PCA降到50维。(2)对照组识别:三级匹配协议——先对'control/ctrl/NT'等关键词做不区分大小写匹配,再用定界符感知正则做短token匹配,最后做子串匹配;Replogle额外处理'non-targeting'/'chr'前缀并剔除'pos_control';Papalexi把NTg1–NTg7合并为单一对照(2386个细胞)。(3)几何稳定性计算:对每个扰动 $p$(≥50个细胞,Dixit阈值为10),每个细胞算 $d_i = x_i - \mu_{ctrl}$,平均方向 $\bar{d}=\frac{1}{n_p}\sum_i d_i$,最后 $S_p = \frac{1}{n_p}\sum_{i=1}^{n_p}\frac{d_i\cdot \bar{d}}{\|d_i\|\|\bar{d}\|}$;效应量 $M_p=\|\bar{d}\|$。(4)非线性discordance:用LOESS(带宽分数0.3)拟合 $M_p$ 与 $S_p$ 关系,残差即为discordance;线性/秩残差作为对照在SI中给出。(5)GO功能多样性验证:对Norman每个扰动取top-25/50/100差异表达基因,g:Profiler做GO:BP富集,统计显著term数。(6)PS三档对比:Tier1=质心欧氏距离,Tier2=Mahalanobis距离,Tier3=Python移植的scMAGeCK约束优化算法;bootstrap 10000次(seed=320)算偏相关。(7)分裂半重复性:对每个扰动随机分两半50次,算两半均值平移向量的余弦相似度均值;在效应量四分位内对比高/低Sp亚组。(8)通路应激评分:scanpy.tl.score_genes计算MSigDB Hallmark四套(UPR 72–78基因、p53 32–51、Apoptosis 53–73、ROS 45–55),与Sp做Spearman偏相关,BH校正。(9)scGPT验证:用'Whole Human'预训练checkpoint对原始counts做embed_data()嵌入,复算Sp和 $M_p$。
技术新颖性
技术新颖性体现在三个方面。第一,度量本身的简洁性:Sp只需一个AnnData对象加扰动标签就能算,作者把它打包成开源Python包shesha-geometry(PyPI),极低使用门槛。相比TRADE需要复杂统计模型、scMAGeCK需要约束优化,Sp是一个透明的单公式指标。第二,对非线性残差的强调:作者明确指出效应量-稳定性关系在低幅度端是非线性的(信噪比约束了连贯性),因此使用LOESS而非线性回归;这一选择让CEBPA+JUN从线性rank 1掉到LOESS rank 172,揭示了高幅度区非线性最严重,避免了'大效应量偏差'导致的高排名假象。第三,对PS基线的批判性重实现:作者没有接受基于质心的距离代理作为PS的简化版,而是移植了scMAGeCK算法重新比较,发现真实PS与质心代理的相关性只有0.097(之前文献中报道的强负相关其实是几何重言式),这种'质疑代理、重做基线'的科研态度在工具类论文里值得借鉴。
实验结果
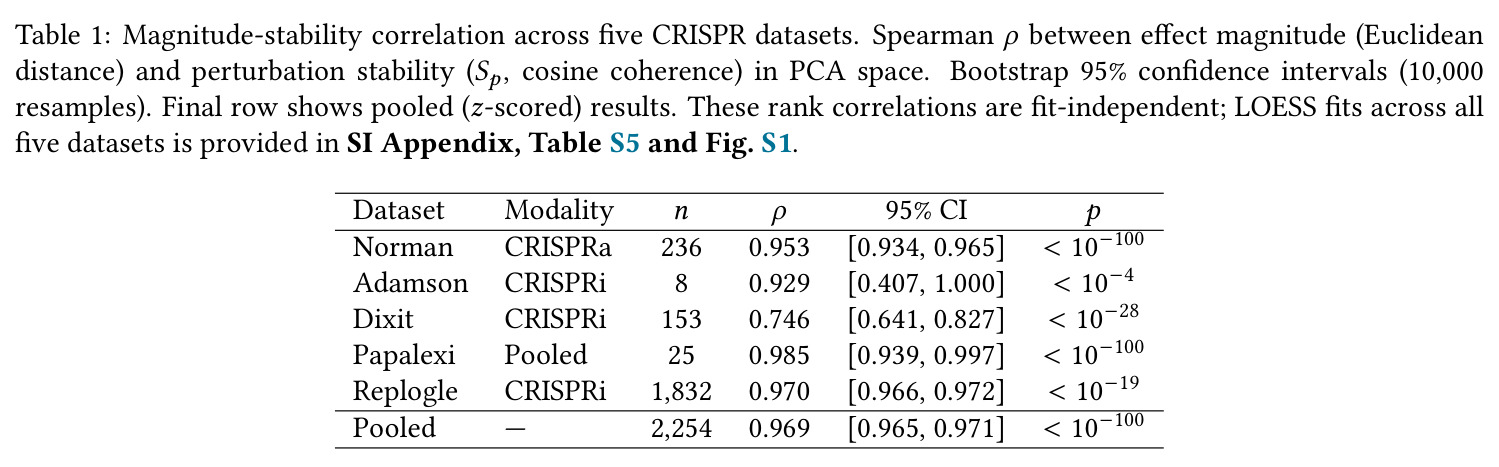
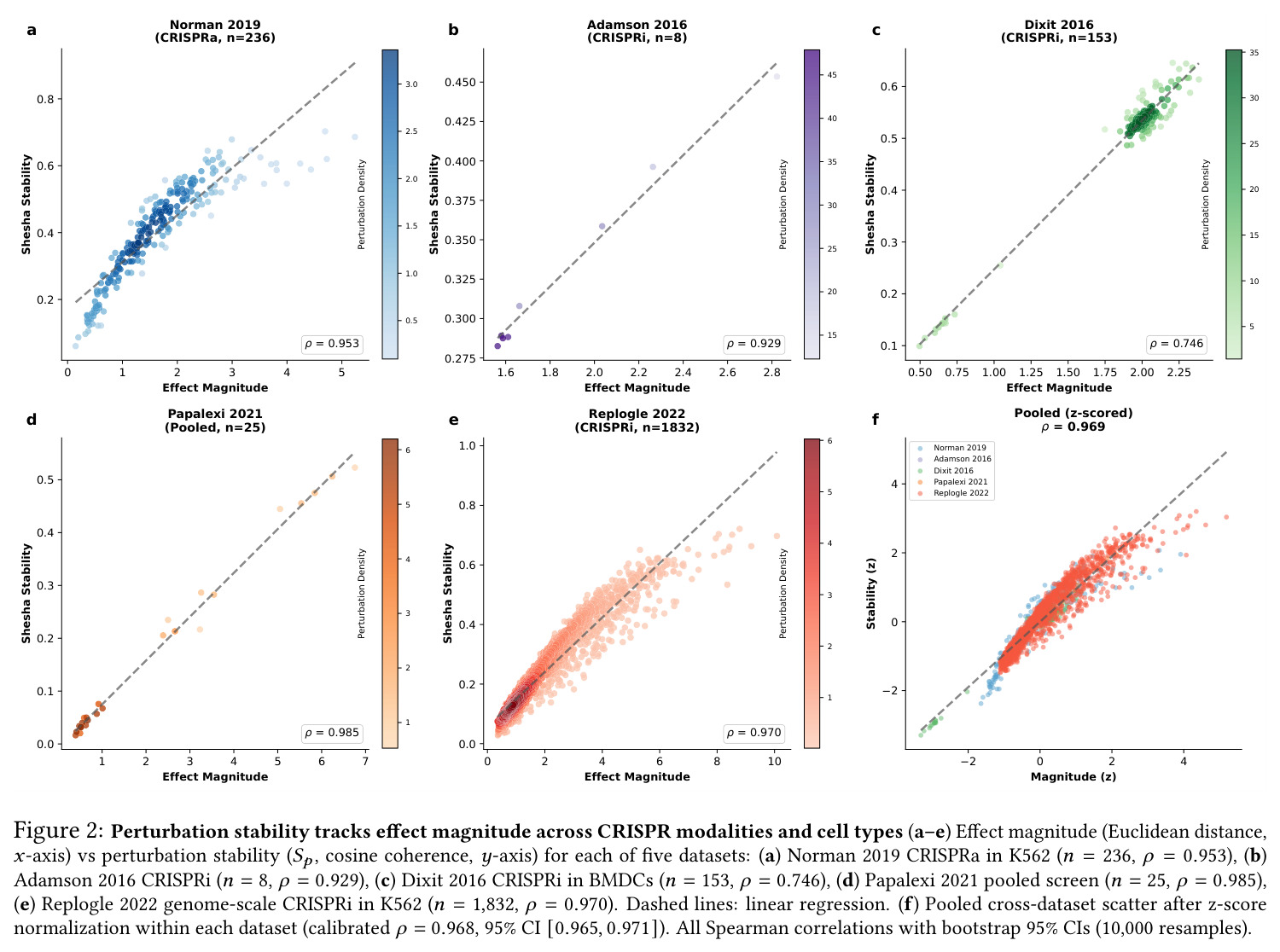
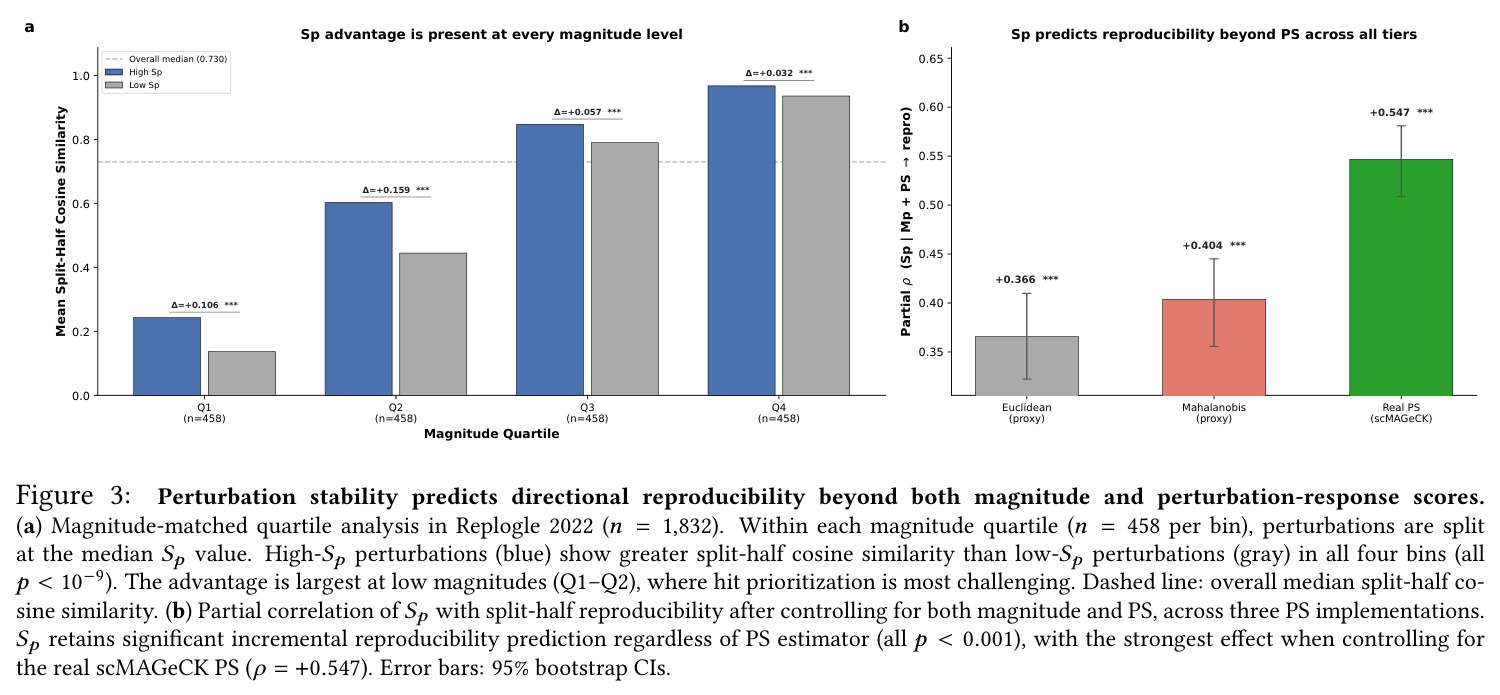
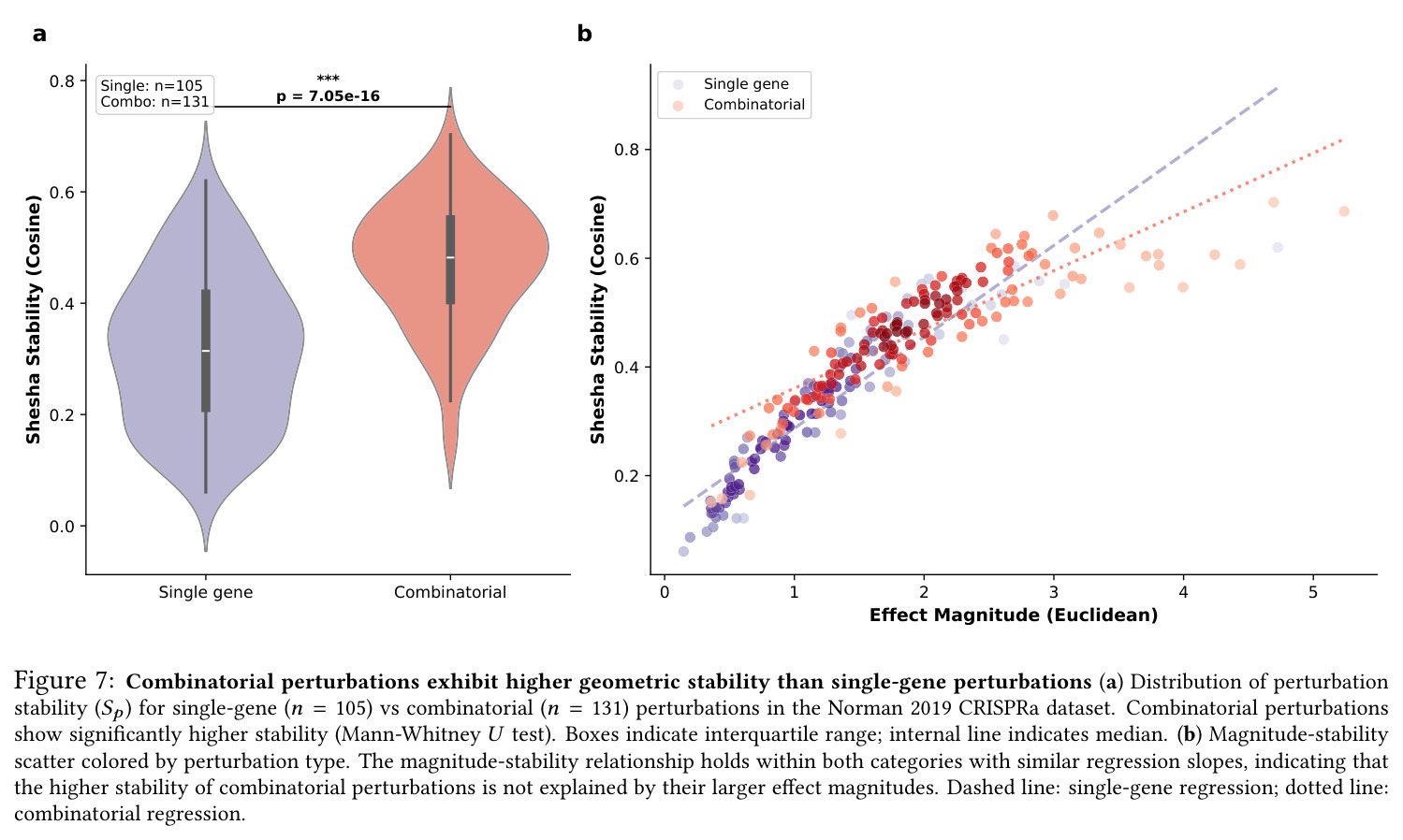
核心发现可归纳为五层。第一,效应量-稳定性强相关跨数据集普适:Spearman $\rho$ 在Dixit 0.746(CI [0.641, 0.827])到Papalexi 0.985之间,五个数据集全部显著(最低 $p<10^{-28}$),合并2254扰动池化 $\rho=0.969$([0.965, 0.971]),混合效应模型显示幅度 $\beta=0.168$ 占解释力约11倍于样本量($\beta_{n\_cells}=-0.015$)。第二,'几何税'在pleiotropic调控因子上明确:Norman中CEBPA家族在线性残差下占discordant极端(CEBPA+JUN rank 1、CEBPA rank 2),LOESS校正后CEBPA+JUN掉到rank 172;KLF1在两种残差下都是最concordant单基因扰动。Replogle上LOESS最discordant的是CHMP2A(5.07)、SF3B3(4.83)、SF3B2(4.55)、PSMD7(4.23)、CHMP3(4.20),全为多亚基复合物亚基。功能多样性:CEBP家族平均21.1个GO:BP term vs KLF1组合9.6个($p=0.013$),但全局不相关($\rho=-0.119$,$p=0.11$)。第三,Sp预测分裂半重复性超越幅度与PS:Replogle控制幅度后Sp偏相关 +0.387($p=1.3\times10^{-66}$),三种PS全部为负(-0.249到-0.184)。在幅度四分位内高Sp亚组在每个bin都更高(Q1 $\Delta=+0.106$ 到 Q4 $+0.032$)。第四,Sp独立预测UPR激活:四个数据集UPR偏相关全部为负(Replogle -0.214、Dixit -0.231、Papalexi -0.395方向一致但样本不足、Norman -0.023),控制幅度和真实PS后Sp仍预测UPR($\rho=-0.203$,$p=1.9\times10^{-18}$),反向PS只有-0.072。HSPA5偏相关在Dixit -0.338、Replogle -0.206最稳定。第五,scGPT非线性嵌入复现关系:Norman 0.935、Dixit 0.712、Replogle 0.851,数据集排名与PCA一致。组合扰动Sp显著高于单基因($p=7\times10^{-16}$),不能由幅度差异解释。
查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| Magnitude-stability correlation (Replogle 2022, n=1832) | Spearman ρ between Euclidean magnitude and cosine Sp | 0.970 (95% CI [0.966, 0.972]) | Magnitude-only framework (Pearson on mean shift) | Quantifies directional coherence layer orthogonal to magnitude |
| Split-half reproducibility (Replogle 2022, controlling for magnitude) | Partial Spearman ρ with cosine similarity of half-shift vectors | +0.387 (95% CI [+0.345, +0.428], p=1.3×10⁻⁶⁶) | Song et al. PS (scMAGeCK): -0.193 (p=7.1×10⁻¹⁷) | Sign flip and ~5x larger magnitude vs PS |
| UPR incremental prediction (Replogle 2022, controlling for magnitude and PS) | Partial Spearman ρ with Hallmark UPR composite score | Sp: -0.203 (95% CI [-0.248, -0.156], p=1.9×10⁻¹⁸) | Real PS: -0.072 (p=2.1×10⁻³) | Sp incremental R² ~3x larger than PS after the same controls |
| HSPA5 partial correlation (Dixit 2016, n=153) | Partial Spearman ρ controlling for magnitude | -0.338 (95% CI [-0.506, -0.164], p=1.9×10⁻⁵) | DDIT3 raw -0.313 vs partial collapsed/reversed | HSPA5 partial survives magnitude control, DDIT3 does not |
| scGPT embedding validation (Norman 2019, n=236) | Spearman ρ between magnitude and Sp in nonlinear embeddings | 0.935 (95% CI [0.911, 0.951]) | PCA: 0.953 | Confirms relationship is intrinsic to biological state space, not linear projection artifact |
| Combinatorial vs single-gene stability (Norman 2019) | Mann-Whitney U on Sp distribution | Combinatorial mean significantly higher, p=7.05×10⁻¹⁶ | Single-gene baseline (mean within-pair regression slope matched) | Combinatorial perturbations engage canalized trajectories more deeply |
局限与改进
作者明确承认的限制包括:(1)PCA是主要嵌入,scGPT验证虽缓解线性投影担忧,但流形感知方法(扩散映射、PHATE)可能揭示更多非线性结构;(2)Adamson数据集仅 $n=8$,bootstrap CI极宽,且pertpy提供的版本是TF knockdown而非原文UPR臂,不能作为应激关联的阳性对照;(3)discordance排名对残差方法敏感,线性vs LOESS在高幅度区差异显著(如CEBPA+JUN从rank 1掉到rank 172),具体基因排名需谨慎解读;(4)应激-稳定性关联仍是相关性而非因果,建立因果需要滴定gRNA递送或诱导系统独立控制扰动效率;(5)Sp是全局标量,无法捕捉亚群结构、双峰响应或剂量依赖异质性;(6)基因级而非guide级分析意味着未测量的guide效率差异可能被误判为incoherence,特别是高幅度扰动常合并多条guide。我自己的额外观察:Norman CRISPRa上Sp-UPR关联近零($\rho=-0.023$)而CRISPRi两个数据集都显著,作者倾向'激活模态衰减'的解释,但样本量236 vs 1832的统计功效差异本身就能解释,文中未做功效分析来区分这两种可能;此外,2,254扰动合并的池化分析未考虑数据集内部异方差,LOESS在跨数据集合并时是否合理缺乏讨论;scGPT PCA的相关系数下降被解释为'非线性嵌入了更多流形结构',但也可能是foundation model对扰动信号的压缩损失,未做对照实验排除。
独立分析的弱点
独立分析的几个弱点及改进方向。第一,应激-稳定性因果链尚未建立:目前Sp低→UPR高的观察无法排除'某些基因类别(分泌通路蛋白、跨膜受体)天然既几何发散又激活UPR'的混淆变量解释。改进方向是设计titrated knockdown实验,用不同强度gRNA或诱导型dCas9独立调控扰动'幅度'与'同步性',再测UPR标志物;若仅在Sp低但幅度受控的扰动下UPR才升高,则支持因果性。第二,Sp对剪接体/ESCRT型incoherence的解读缺乏分子机制深挖:作者把这些亚基归为'异质性部分功能丧失'但未定量刻画残余复合物活性在不同细胞中的分布。改进方向是用单分子荧光原位杂交对SF3B3敲除细胞做剪接体装配状态单细胞读出,与Sp做联合分析,从机制层面验证'残余复合物活性差异→转录组方向发散'链条。第三,2,254扰动合并的discordance排名本质是回归残差,无法识别'幅度中等但方向极端'的稀有扰动;Norman中CEBPA+JUN从rank 1掉到LOESS rank 172正是被高幅度端非线性吃掉。改进方向是开发基于非参数密度估计的discordance定义,让'方向极端'独立于幅度成为度量目标。第四,组合扰动Sp更高的结论受Norman配对设计的选择偏倚影响,不能推广到随机配对,需分析现有随机配对数据集作为对照。第五,Sp仅在PCA/scGPT嵌入中验证,未在蛋白质组、ATAC-seq或空间转录组层面检验'方向一致性'是否跨模态稳健,改进方向是把Sp推广到多模态单细胞数据。
未来方向
作者提出的方向包括:滴定扰动系统独立控制幅度与同步性以建立因果;空间分辨Perturb-seq(如Nevue et al. 2026的PerturbSpace)测试incoherence是否受组织微环境调节;CD4+ T细胞原代细胞图谱上测试'几何税'是否在原代细胞中重现;Sp作为in silico扰动预测工具(GEARS、CellOracle、CPA)的辅助评估指标。我基于成果可延伸的方向:(1)把Sp与方向传输图或最优传输理论结合,开发'方向流场'可视化,让每个扰动的群体响应不只是标量而是一个PCA空间中的向量场,更直观地揭示分歧轨迹;(2)结合scVI/scGPT等基础模型的隐空间几何(如局部曲率、Ricci曲率)作为协变量,分析Sp与流形曲率的耦合关系,从'方向一致性'升级到'曲率地形学';(3)把Sp与因果基因调控网络推断结合:低Sp基因往往是多靶点TF或复合物亚基,可作为GRN推断算法中'hub regulator'的独立信号;(4)开发时间分辨Perturb-seq数据集的时序版本Sp——观察细胞是否在时间维度上也保持方向一致,把几何稳定性从静态扩展到动态景观。
复现评估
复现性总体良好。开源代码:作者把Sp实现打包成shesha-geometry Python包并发布到PyPI,使用门槛极低——只需一个带扰动标签和控制条件的AnnData对象即可计算,无需GPU。数据集:五个数据集全部通过pertpy 1.0.4公开访问,无授权问题;唯一例外是Adamson $n=8$在pertpy中只有TF knockdown版本而非原UPR臂,限制了UPR阳性对照的复现。算力需求:PCA + 余弦相似度的Sp计算预计单机CPU几分钟内完成;scGPT验证需要GPU嵌入(中等显存),可选'Whole Human'预训练checkpoint降低下载量。关键超参数都已报告:PCA维度50、control匹配三级协议、阈值50细胞(Dixit 10)、LOESS带宽0.3、bootstrap 10000次seed 320、分裂半50次随机分割。复现难度:中等偏低——核心公式透明、协议详细,但需注意discordance排名依赖残差方法(线性 vs LOESS结果差异显著,需按主文用LOESS),scMAGeCK Python移植实现细节未在主文给出。一个熟悉scanpy的单细胞研究者可在1-2天内复现主图表。
论文图表
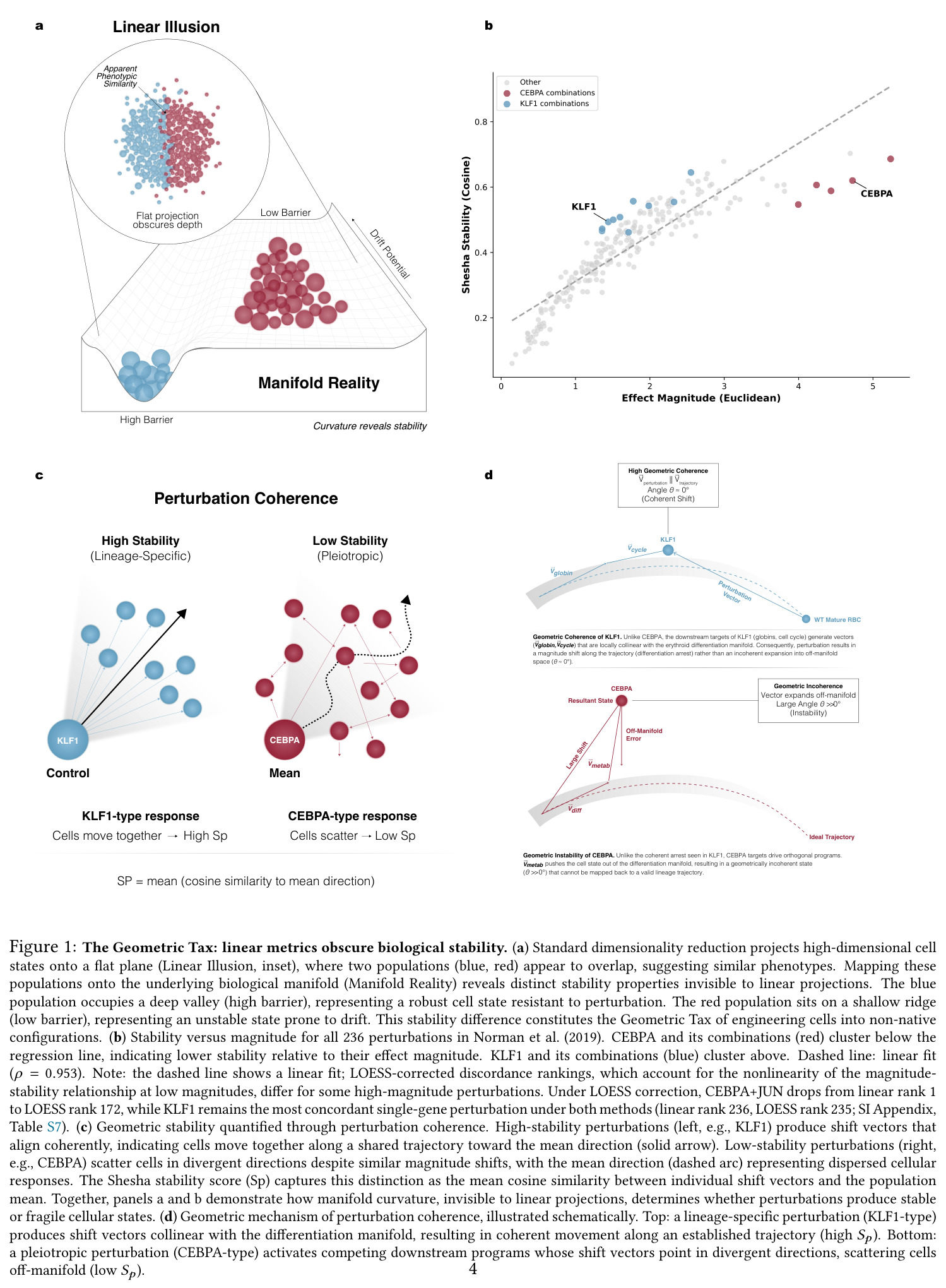
分四个子图展示线性度量如何掩盖生物学稳定性。(a) 线性幻象与流形现实:标准降维把高维状态压到平面,看似重叠的两个人群在底层流形上分别位于深谷(高能垒、稳定)和浅脊(低能垒、易漂移),稳定性差异即'几何税'。(b) Norman数据集236个扰动的幅度-稳定性散点,CEBPA组合(红)聚集在回归线下方、KLF1组合(蓝)聚集在上方,Spearman ρ=0.953。(c) 几何稳定性的量化:KLF1型响应所有平移向量对齐,CEBPA型响应向量发散,Sp即平均余弦相似度。(d) 几何机制示意:KLF1目标与分化流形共线→细胞沿轨迹一致性移动;CEBPA目标驱动正交程序→细胞散落到流形外。
这是论文的'门面图',把抽象的'几何税'概念可视化——没有这张图读者无法直观理解Sp在PCA空间的几何含义以及为什么线性降维会抹掉这种结构。它是后续所有图表的概念基础。