GPCR-Filter:面向高效精准GPCR调节剂发现的深度学习框架 GPCR-Filter: a deep learning framework for efficient and precise GPCR modulator discovery
ESM-3+GNN+注意力融合框架,精准预测GPCR-配体调节关系
前置知识
G蛋白偶联受体(GPCR)
GPCR是细胞表面最大的受体超家族,包含800多个成员,通过偶联G蛋白和β-arrestin调控神经系统、内分泌系统和免疫系统的关键信号通路。GPCR具有高度动态的构象景观和变构机制,配体结合与下游信号效能之间存在解耦现象。美国FDA批准的药物中约36%以GPCR为靶点,目前有超过300个GPCR靶向药物处于临床开发阶段。理解GPCR的生物学特性对于药物发现至关重要。
GPCR是本文研究的核心靶点家族,理解其生物学复杂性是理解模型设计动机和评估指标意义的前提。
ESM-3蛋白质语言模型
ESM-3是由Meta AI开发的大规模预训练蛋白质语言模型,通过在数十亿蛋白质序列上进行自监督学习,能够将氨基酸序列编码为高维的逐残基(per-residue)嵌入表示。这些嵌入捕获了序列中的进化保守性、结构信息和功能特征,无需实验测定的三维结构即可提供丰富的蛋白质表征。ESM-3的输出维度为1536,为下游任务提供了高质量的特征基础。
GPCR-Filter使用ESM-3作为GPCR序列编码器,是模型获取高质量受体表征的关键组件,直接影响模型对GPCR序列决定因素的捕获能力。
图神经网络(GNN)与GCN
图神经网络是一类专门处理图结构数据的深度学习模型。在分子表示中,原子作为节点、化学键作为边构成分子图。图卷积网络(GCN)通过聚合邻居节点特征来更新节点表示,每层操作可表示为 $x_i^{(l+1)} = \sigma(\sum_{j \in N(i)} \frac{1}{c_{ij}} W^{(l)} x_j^{(l)})$,其中 $W^{(l)}$ 为可训练权重矩阵,$\sigma$ 为ReLU激活函数,$c_{ij}$ 为度归一化项。GCN能够捕获局部化学环境和分子拓扑结构。
GPCR-Filter使用单层GCN对配体SMILES转换的分子图进行编码,是模型理解配体化学结构的核心模块。
交叉注意力机制(Cross-Attention)
交叉注意力是Transformer架构中的关键机制,允许一个序列(query)从另一个序列(key/value)中聚合相关信息。在GPCR-Filter中,配体作为query,GPCR残基作为key/value,注意力权重 $\gamma_{ij} = \frac{\exp(x'_{d,i} W_q \cdot x'_{t,j} W_k^T)}{\sum_{j'} \exp(x'_{d,i} W_q \cdot x'_{t,j'} W_k^T)}$ 反映配体原子对GPCR残基的关注程度。这种机制使模型能够动态整合受体和配体的功能相关信息。
交叉注意力是GPCR-Filter的核心融合机制,也是模型可解释性的基础——通过分析注意力权重可以定位关键结合位点残基。
药物-靶点相互作用(DTI)预测
DTI预测是计算药理学的核心任务,旨在从化合物化学结构和蛋白质序列预测两者是否存在相互作用。传统方法包括基于分子对接的结构方法和基于序列的机器学习方法。序列方法的优势在于可利用更大的数据集,但难以捕获GPCR特有的药理学敏感性。代表性模型包括ConPLex(使用对比学习)和TransformerCPI2.0(使用BERT编码器)。
GPCR-Filter被定位为专门针对GPCR的DTI模型,理解通用DTI方法的局限性是理解本文创新点的关键。
研究动机
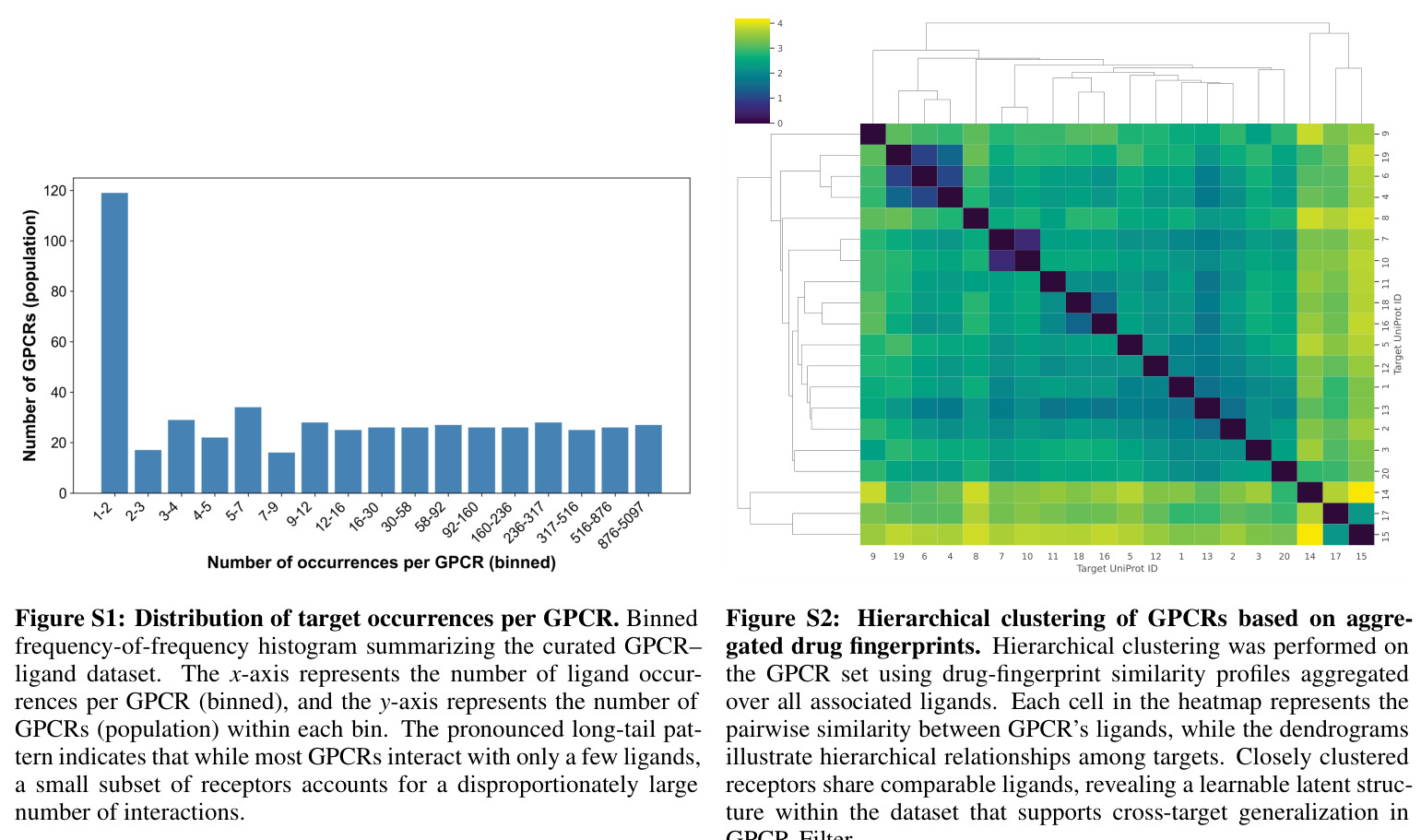
GPCR调节剂的发现面临多重挑战。首先,GPCR的激活往往源于复杂的变构效应而非直接结合亲和力,传统计算筛选方法难以捕获结合与功能调节之间的复杂关系,导致筛选命中率低且存在大量不确定或无活性的假阳性结果。其次,实验测定的GPCR-配体复合物结构数量有限(少于1800个,且高度保守),计算预测结构的不准确性进一步限制了基于结构的方法的有效性。第三,现有的通用DTI模型如ConPLex和TransformerCPI2.0主要针对通用结合预测优化,缺乏捕获GPCR特异性序列-结构-功能关系所需的生化敏感性。此外,实验验证方法成本高昂、通量低、劳动密集,进一步限制了新型调节剂的发现速率。在数据层面,GPCR-配体相互作用数据呈现显著的长尾分布:少数高度混杂的GPCR(如松弛素受体1有5097条记录)主导数据集,而大多数受体仅有稀疏的配体覆盖。
本文的目标是本文的具体目标是开发一个专门针对GPCR调节剂发现的深度学习框架GPCR-Filter,该框架能够仅基于GPCR氨基酸序列和配体SMILES字符串预测调节潜力,作为传统结构筛选之后的二次过滤器。具体而言,研究团队旨在:(1)构建大规模高质量的GPCR-配体相互作用数据集,整合GPCRdb和GtoPdb的实验验证记录;(2)设计融合蛋白质语言模型和图神经网络的多层架构,捕获GPCR与配体之间的功能关系;(3)在随机划分、靶内划分和跨靶划分三种评估场景下全面评估模型泛化能力;(4)通过湿实验验证模型预测能力,确认其在真实药物发现中的实用价值。
与已有工作不同的是,本文的独特切入角度在于三个方面。第一,与通用DTI模型不同,GPCR-Filter专门为GPCR家族设计,针对该家族特有的长尾数据分布、变构调节机制和功能多样性进行了定制化建模。第二,模型创新性地将ESM-3预训练蛋白质语言模型与图神经网络相结合,通过基于注意力的融合机制学习受体-配体功能对应关系,而非仅仅预测结合可能性。第三,研究设计了从易到难的三级评估体系(随机划分→靶内划分→跨靶划分),系统评估模型在不同分布偏移下的泛化能力,并通过可解释性分析验证模型是否真正学到了转移性结合原理而非简单记忆。这种从数据组织、模型设计到评估策略的系统性创新,填补了GPCR领域缺乏专用高效筛选工具的空白。
核心方法
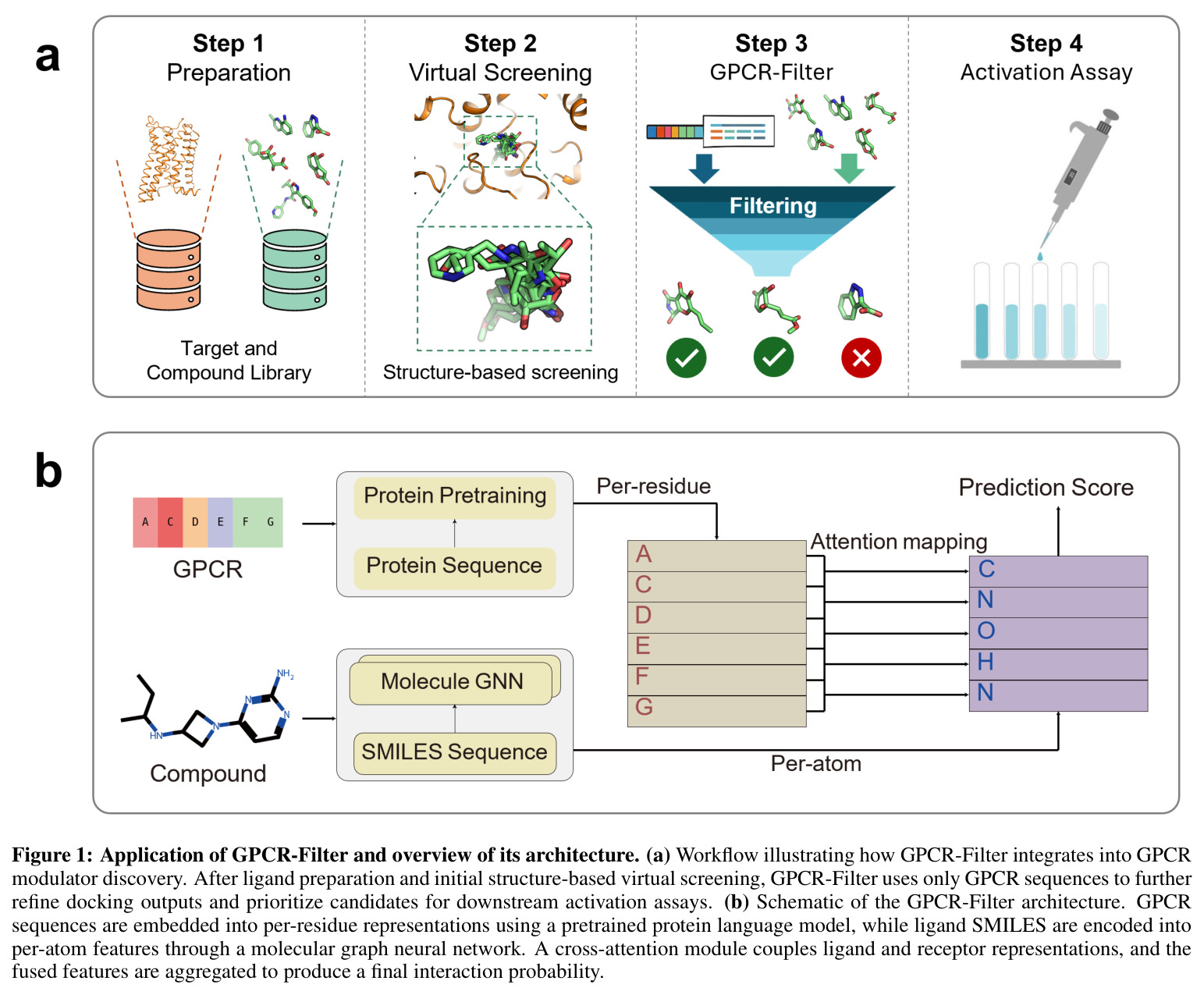
GPCR-Filter的整体思路是将GPCR调节剂发现建模为序列级别的二分类问题:给定GPCR氨基酸序列和配体SMILES字符串,预测两者之间是否存在调节性相互作用。直觉上,模型需要同时理解受体的序列决定因素(哪些残基参与配体识别)和配体的化学结构特征(哪些原子/官能团介导相互作用),并将两者在共享表示空间中进行融合以学习功能对应关系。技术路线上,GPCR侧使用ESM-3预训练模型提取逐残基嵌入,配体侧使用RDKit将SMILES转换为分子图后通过GCN提取逐原子特征,两者通过线性投影对齐到共享隐藏维度 $d=256$,最后通过Transformer风格的解码器(配体到蛋白质的交叉注意力)进行特征融合,输出交互概率。这种设计使模型能够在保留各自领域专业知识的同时,通过注意力机制动态发现受体和配体之间的功能关联。
GPCR-Filter的核心创新点在于其多层融合架构设计,与已有方法存在本质区别。首先,与ConPLex使用对比学习在蛋白质语言空间中预测相互作用不同,GPCR-Filter采用显式的交叉注意力机制,使配体token能够直接关注GPCR残基,从而获得更具可解释性的交互模式。其次,与TransformerCPI2.0使用通用BERT编码器不同,GPCR-Filter利用ESM-3这一更强大的蛋白质语言模型(1536维嵌入),能够捕获更精细的序列进化信息。第三,解码器采用配体到蛋白质的单向交叉注意力(而非双向),从配体的图级CLS token($t=0$)作为query聚合GPCR残基信息,这种设计更符合物理直觉——配体需要寻找受体上的结合位点。最后,模型将预测读数从图级token(序列第一个位置)提取,使得最终预测聚焦于整体分子级别的相互作用而非局部原子级别。
方法步骤详情
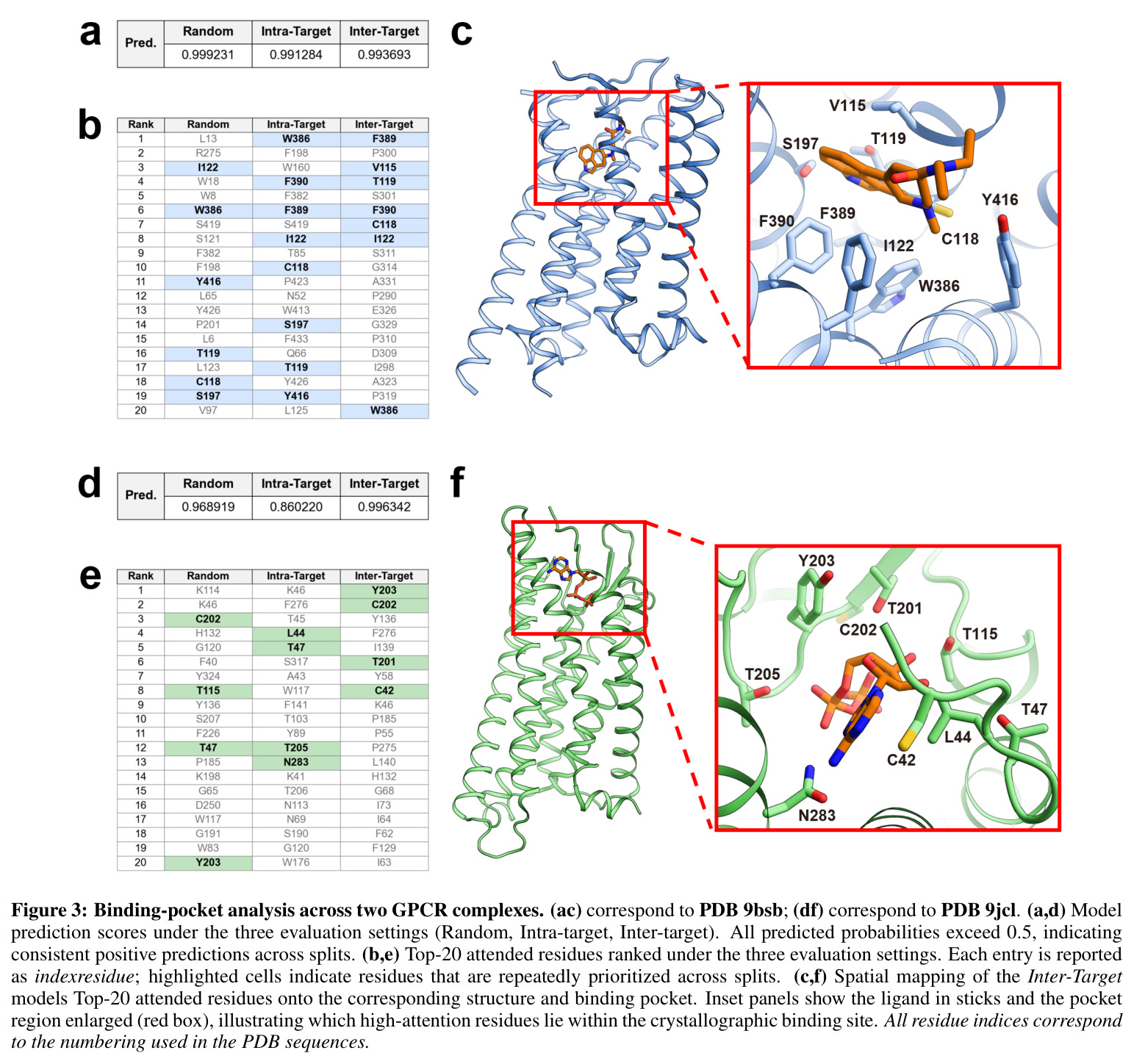
GPCR-Filter的方法包含以下完整步骤。第一步,GPCR嵌入计算:从ESM-3获取维度为 $h_t=1536$ 的逐残基表示,通过线性投影 $R^{h_t} \rightarrow R^d$ 映射到共享隐藏维度 $d=256$,然后通过 $L=2$ 层Transformer编码器(8头注意力,前馈宽度 $4d$,dropout 0.1)进行序列编码,编码后的序列作为解码器的记忆。第二步,配体嵌入计算:使用RDKit将SMILES转换为分子图 $G=(V,E)$,节点特征经线性投影后通过单层GCN卷积,然后在原子序列前添加可学习的图级token(用于图级读取),并填充到批次最大长度形成目标序列 $\tilde{X}_d \in R^{(1+|V|) \times d}$。第三步,解码器与注意力融合:配体序列作为target/query,编码后的蛋白质序列作为memory/key-value。首先配体进行自注意力以精炼表示,蛋白质也进行编码器自注意力;然后配体作为query、蛋白质作为key/value进行交叉注意力,更新配体特征 $x''_{d,i} = \sum_j \gamma_{ij}(x'_{t,j}W_v) + x'_{d,i}$。第四步,预测输出:提取图级token $x''_d[0] \in R^d$,通过MLP映射为二分类logits,使用softmax计算正类概率 $p = \text{softmax}(o)[1]$。第五步,训练目标:最小化二元交叉熵损失 $\mathcal{L} = -\sum_i \log p(y_i | x_i)$。第六步,可解释性分析:提取最后一层交叉注意力权重,从配体图级token到残基token,跨头平均,L1归一化后排序得到Top-20重要残基。
技术新颖性
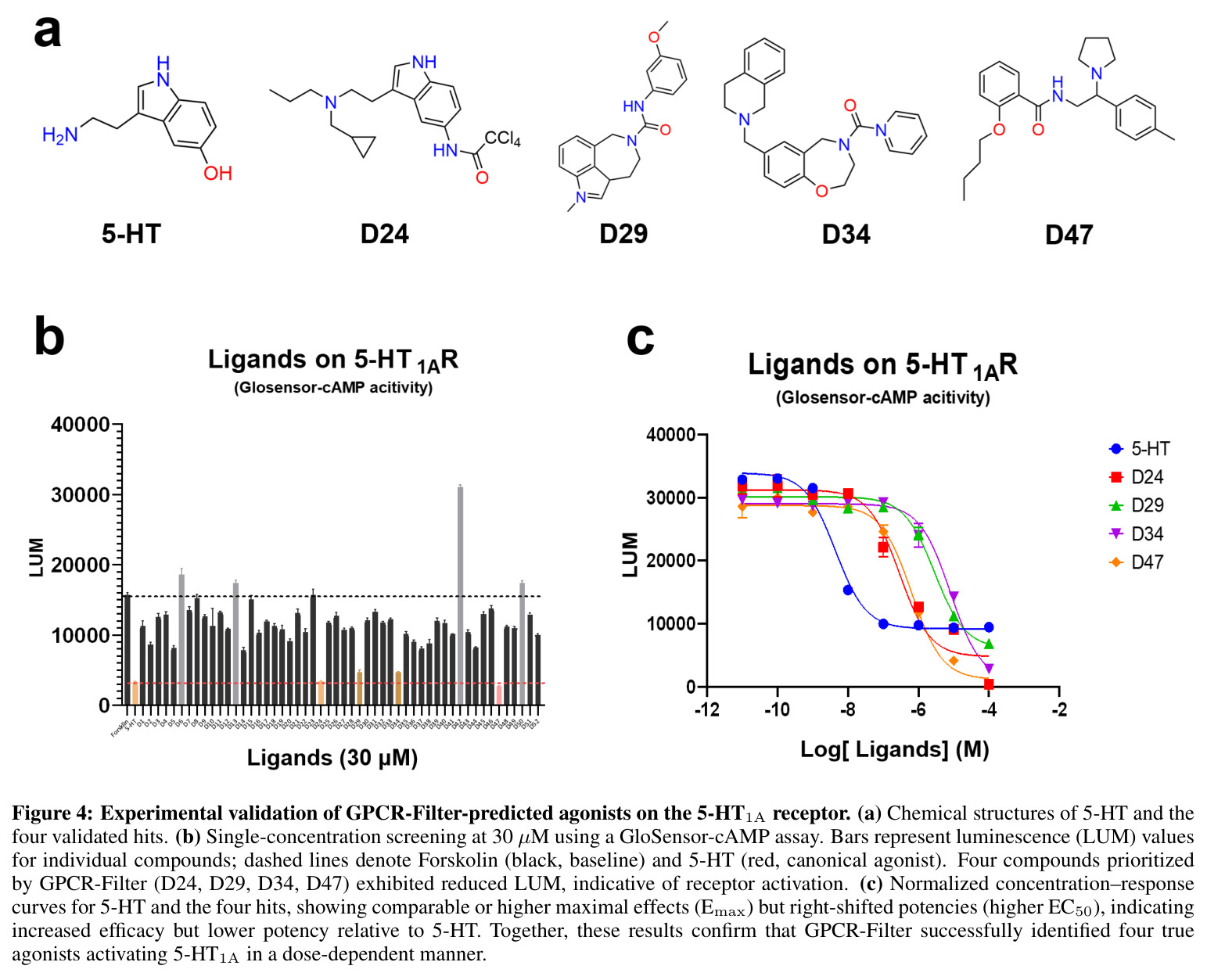
GPCR-Filter的技术新颖性体现在多个层面。在数据层面,研究团队构建了包含91,396条实验验证的GPCR-配体相互作用数据集,涵盖527个独特GPCR和72,177个独特配体,这是该领域最大规模的高质量数据集之一。通过系统化的负采样策略(枚举所有组合、去除已知正样本、1:1平衡采样)构建训练数据。在模型架构层面,创新性地将ESM-3蛋白质语言模型与GCN结合,通过配体到蛋白质的单向交叉注意力实现特征融合,这种设计既保留了预训练模型的表征能力,又通过注意力机制实现了可解释的交互建模。在评估策略层面,设计了从易到难的三级评估体系:随机划分测试分布内性能,靶内划分测试同一受体的新配体泛化,跨靶划分测试全新受体的迁移能力。这种系统化的评估框架比单一划分策略更能全面评估模型能力。在验证层面,通过湿实验在5-HT1A受体上验证了4个微摩尔级别激动剂,证明模型的实用价值。
实验结果
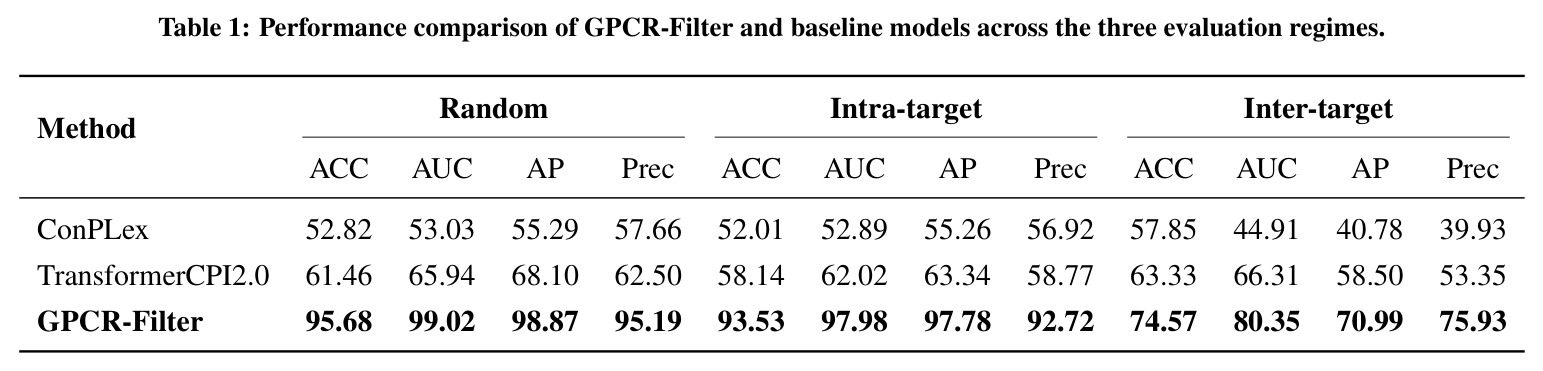
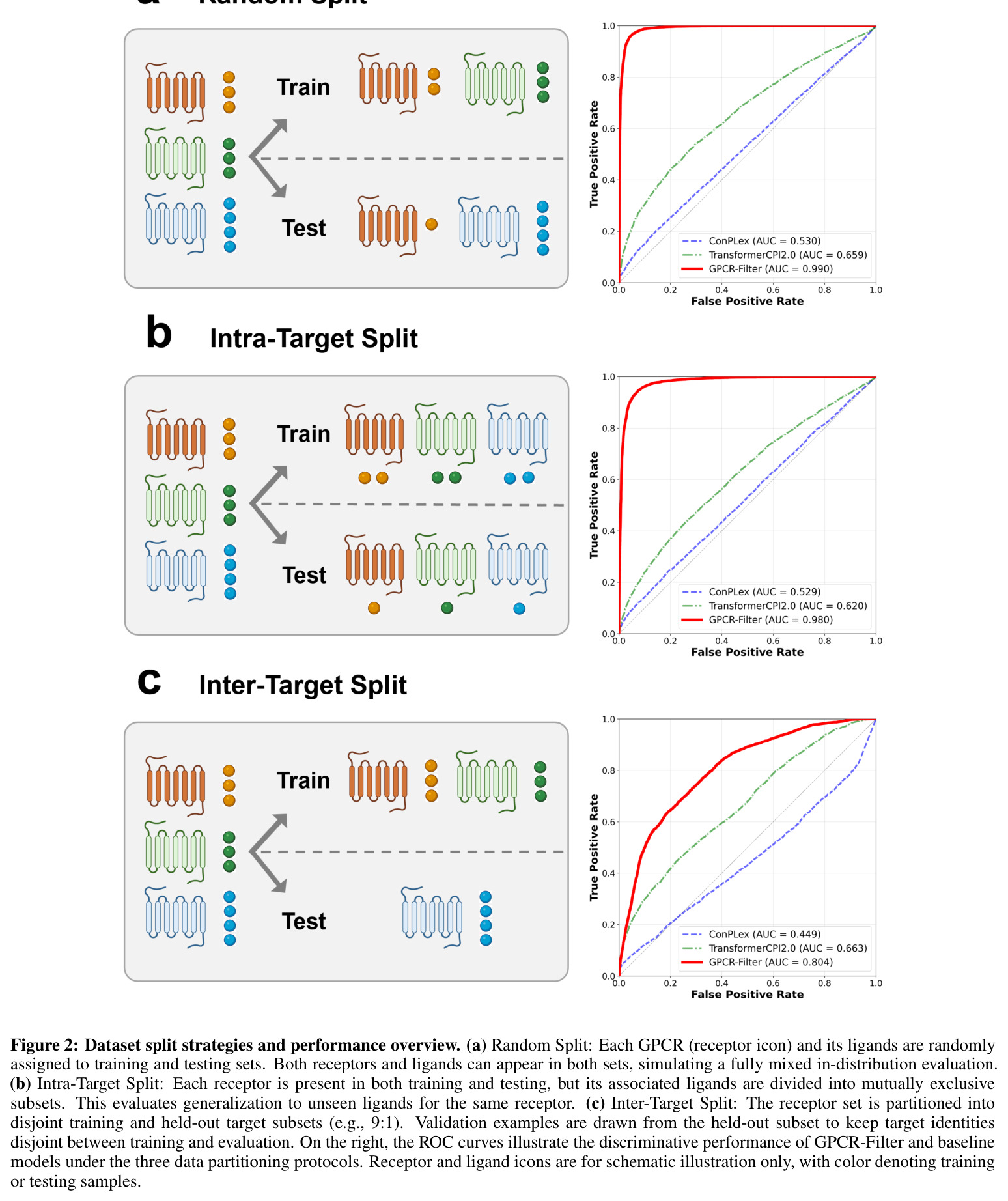
GPCR-Filter在三种评估场景下均显著超越基线模型。在随机划分(分布内评估)中,GPCR-Filter达到AUC 99.02%和AP 98.87%的接近天花板性能,而ConPLex(AUC 53.03%)和TransformerCPI2.0(AUC 65.94%)表现远逊。在更具挑战性的靶内划分中(测试同一受体的新配体组合),GPCR-Filter保持AUC 97.98%和AP 97.78%,而两个基线模型AUC均低于63%,表明其对配体空间的泛化能力有限。在最严格的跨靶划分中(测试完全未见过的受体),GPCR-Filter达到AUC 80.35%和AP 70.99%,显著优于TransformerCPI2.0(AUC 66.31%)和ConPLex(AUC仅44.91%,低于50%说明过拟合)。可解释性分析显示,Top-20注意力排名残基在两种GPCR复合物(DRD2 PDB 9bsb和嘌呤能受体PDB 9jcl)中均富集于晶体学口袋残基,随机、靶内和跨靶模型分别回收了6/9、8/9、7/9和类似比例的口袋残基,证明模型学到了转移性结合原理。湿实验验证成功在5-HT1A受体上鉴定出4个微摩尔级别激动剂(D24、D29、D34、D47),其最大效应(Emax)接近或超过5-HT,但效价较低(EC50约微摩尔级),表明这些化合物是有前景的亲和力优化起点。
查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| 随机划分(分布内评估) | AUC | 99.02% | ConPLex 53.03%, TransformerCPI2.0 65.94% | 较最优基线提升33.08个百分点 |
| 随机划分(分布内评估) | AP | 98.87% | ConPLex 55.29%, TransformerCPI2.0 68.10% | 较最优基线提升30.77个百分点 |
| 靶内划分(新配体泛化) | AUC | 97.98% | ConPLex 52.89%, TransformerCPI2.0 62.02% | 较最优基线提升35.96个百分点 |
| 靶内划分(新配体泛化) | AP | 97.78% | ConPLex 55.26%, TransformerCPI2.0 63.34% | 较最优基线提升34.44个百分点 |
| 跨靶划分(全新受体迁移) | AUC | 80.35% | ConPLex 44.91%, TransformerCPI2.0 66.31% | 较最优基线提升14.04个百分点 |
| 跨靶划分(全新受体迁移) | AP | 70.99% | ConPLex 40.78%, TransformerCPI2.0 58.50% | 较最优基线提升12.49个百分点 |
局限与改进
作者承认的局限性包括:第一,负标签通过1:1负采样获得,不可避免地包含未观察到的真正阳性样本,探索替代decoy策略、采样比例和重采样方差可能进一步明确性能边界。第二,在跨靶划分中,配体被允许在不同目标间重复出现;虽然受体本身是不相交的,但配体复用可能为某些化学型提供更简单的信号,更严格的配体或支架级去重可以进一步压力测试模型。第三,更广泛的比较涉及更深入的校准策略或结构感知模型仍有待探索。从独立观察来看:随机划分的接近天花板性能(AUC约0.99)可能部分反映了随机划分的记忆倾向性和潜在的数据泄漏;跨靶划分中ConPLex的AUC低于50%但准确率高于50%,作者将其归因于过拟合,但这也可能反映了评估协议的某些特性;数据集的长尾分布意味着模型在稀有GPCR上的性能可能不如在高频GPCR上,但论文未提供按GPCR频率分层的性能分析。
独立分析的弱点
GPCR-Filter存在几个值得改进的弱点。首先,模型仅使用单层GCN对配体进行编码,这可能限制了对复杂分子结构的捕获能力;对于具有长程依赖的分子(如大环化合物),更深的图网络或图注意力网络(GAT)可能更有效。其次,负采样策略采用简单的1:1平衡采样,未考虑不同GPCR的先验活性率差异;在实际药物发现中,活性化合物比例通常远低于1:1,模型在更现实的不平衡场景下的性能未知。第三,模型的可解释性分析仅限于定性的注意力权重可视化,缺乏定量的统计检验和与已知药效团模型的系统比较。第四,湿实验验证仅在单一受体(5-HT1A)上进行,且仅验证了4个化合物,样本量较小,难以全面评估模型的筛选能力。改进方向包括:引入更复杂的负采样策略(如基于化学多样性的采样)、增加图网络深度、在更多GPCR亚型上进行实验验证、以及开发定量的可解释性评估指标。
未来方向
作者提出的未来工作方向包括:(1)扩展验证到更多蛋白质家族,建立GPCR-Filter在更广泛靶点上的适用性;(2)改进负采样和校准协议,探索替代decoy策略和采样比例;(3)纳入结构或口袋级约束以进一步增强跨靶泛化能力。基于已有成果可延伸的研究方向包括:将GPCR-Filter与结构基虚拟筛选方法(如分子对接)进行更深入的集成,形成端到端的筛选流水线;利用注意力机制发现的口袋残基信息指导配体优化;探索迁移学习在不同GPCR家族间的应用(如从A类GPCR迁移到B/C类);开发生成式模型基于GPCR-Filter的评分函数进行配体从头设计;以及将模型扩展到多靶点GPCR调节剂的同时预测。
复现评估
论文提供了较为详细的实验设置描述,包括模型架构参数($d=256$, $L=2$, 8头注意力,前馈宽度$4d$,dropout 0.1)、训练细节(二元交叉熵损失,固定阈值0.5)和评估协议(80/10/10划分比例)。数据集来源于公开数据库(GPCRdb和GtoPdb),理论上可复现。然而,论文未明确说明是否开源代码和预训练模型权重,这对完全复现至关重要。ESM-3作为预训练模型需要较大的计算资源(GPU内存和推理时间),这可能对资源有限的研究者构成门槛。湿实验验证部分使用了商业化合物库(ChemDiv,1,644,833个化合物),这些化合物的获取成本和可用性可能限制其他团队的验证能力。总体而言,在有足够计算资源和数据访问权限的情况下,模型的核心训练和评估流程应该是可复现的。
论文图表
图S2展示了对GPCR集合进行的层次聚类,使用聚合的药物指纹相似性配置文件。热图中的每个单元格表示GPCR药物配置文件之间的配对相似性,树状图展示了靶点之间的层次关系。化学调节剂相似的受体聚集在一起,揭示了数据集内可学习的潜在结构,支持GPCR-Filter的跨靶泛化能力。
这张图从数据层面解释了为什么跨靶泛化是可能的——GPCR在配体化学空间中存在有组织的结构,为模型学习可转移的化学模式提供了基础。