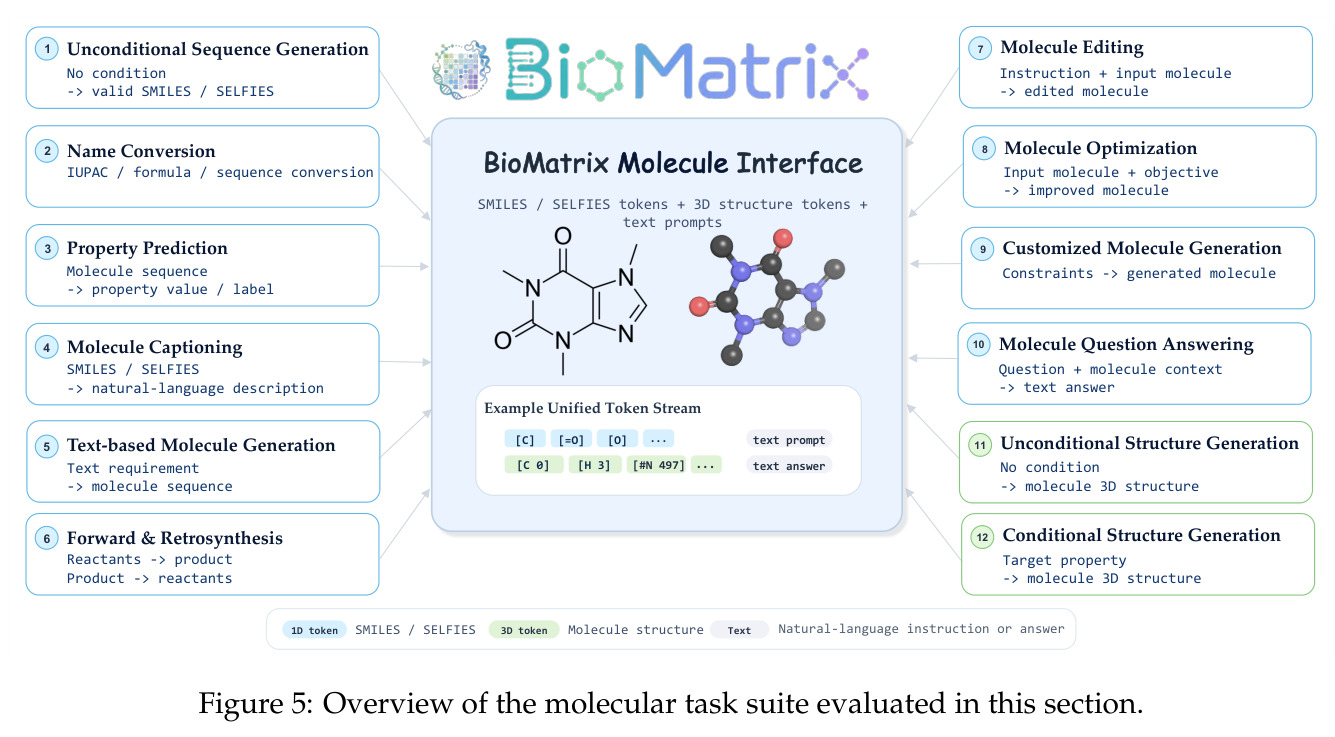
BioMatrix:跨越序列、结构和语言模态矩阵的全面生物基础模型 BioMatrix: Towards a Comprehensive Biological Foundation Model Spanning the Modality Matrix of Sequences, Structures, and Language
首个统一分子与蛋白质的多模态生物基础模型,原生支持序列、结构和语言
前置知识
SMILES/SELFIES
SMILES 是一种用 ASCII 字符串表示分子结构的线性符号,每个重原子对应一个字符,键和环用符号标记。SELFIES 是 SMILES 的改进版本,通过引入分支和环的控制标记,保证任何语法上正确的字符串都能解码为化学上有效的分子,解决了 SMILES 容易生成无效分子的问题。
理解这两种分子表示法对评估 BioMatrix 的分子生成任务至关重要,论文证明了它们在不同任务上的互补性:SELFIES 擅长保证化学有效性,SMILES 擅长结构约束任务。
SE(3)-invariant 结构tokenizer
SE(3) 表示三维欧几里得变换群即旋转和平移。SE(3)-invariant 指的是对物体的整体旋转和平移保持不变的表示。在分子和蛋白质结构中,这意味着无论分子在空间中如何旋转或平移,其内部表示应该相同。论文中使用局部球坐标系和相对几何描述来实现这种不变性。
这是 BioMatrix 将连续的 3D 结构离散化为 token 的关键技术,理解它才能明白为什么模型可以用统一的目标函数处理文本和结构。
向量量化 VQ-VAE
VQ-VAE 是一种将连续编码器输出映射到有限离散码本的技术。编码器将输入转换为连续向量,量化器找到码本中最接近的向量作为离散表示,解码器从离散表示重建原始输入。论文中分子结构使用 512 条目码本,蛋白质结构使用 4096 条目码本。
VQ 是 BioMatrix 将连续的几何信息离散化为语言模型可处理的 token 的核心机制,理解它有助于评估模型在精细几何任务上的局限性。
Residue-level 对齐
蛋白质的结构表示不是按原子粒度,而是按残基粒度对齐的。每个氨基酸残基对应一个序列 token 和一个结构 token,两者一一对应。这意味着序列的第 i 个氨基酸的结构信息由第 i 个结构 token 携带,折叠和反向折叠变成了 token 到 token 的对齐问题。
这是 BioMatrix 能够用统一的 next-token 预测目标同时支持蛋白质折叠和反向折叠的关键设计,与需要特殊架构的专门模型形成对比。
Continual Pretraining
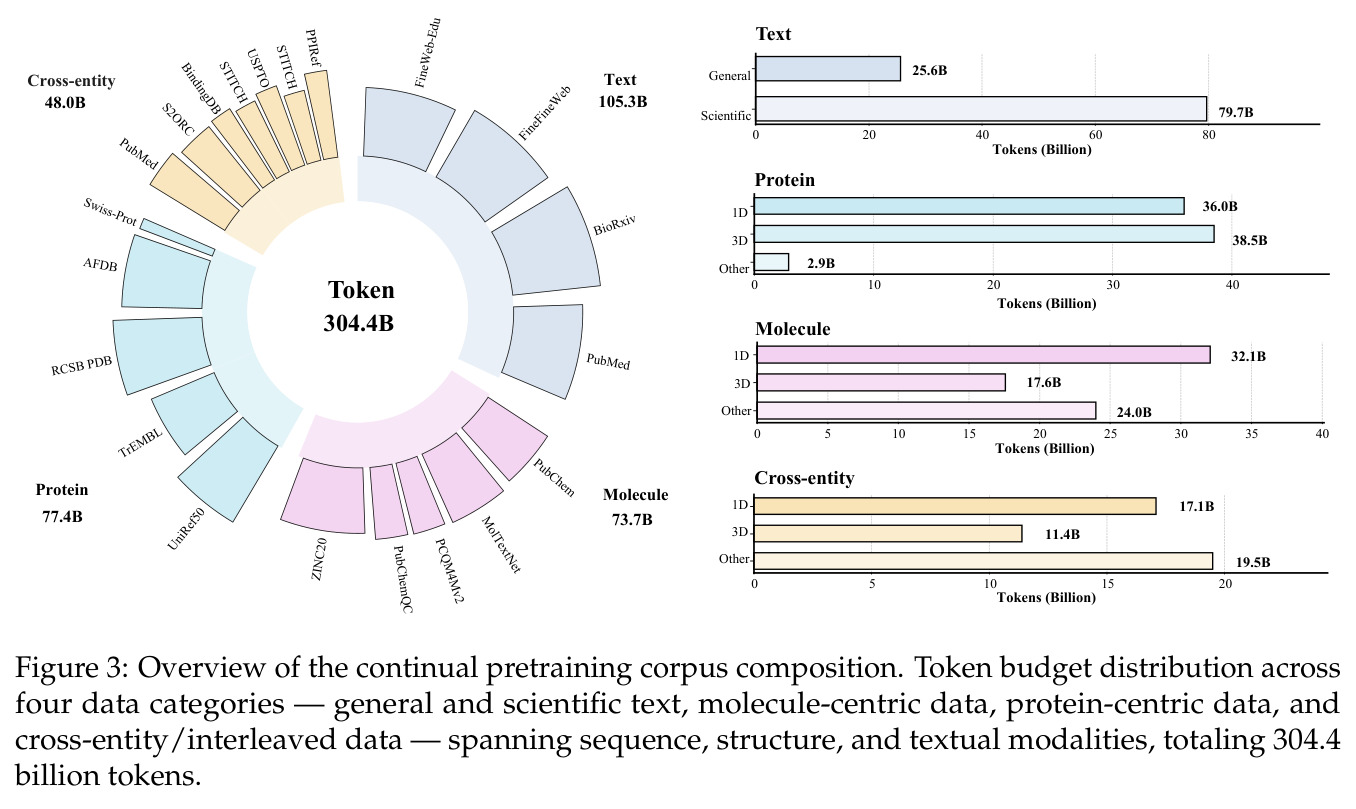
持续预训练是指在已经预训练好的基础模型上,使用特定领域的数据继续训练,而不是从头开始。BioMatrix 在 Qwen3 的基础上,使用 3044 亿 token 的生物医学数据进行了持续预训练,包括通用文本、科学文献、分子和蛋白质数据以及跨实体交互数据。
理解这一点可以评估模型的训练成本和可复现性,以及为什么模型仍保留了部分通用语言能力。
研究动机
现有的生物基础模型在两个维度上存在分离:一是多模态整合能力,二是实体覆盖范围。支持原生多模态(序列、结构、语言统一处理)的模型如 ESM3 仅覆盖蛋白质,而覆盖多实体(分子、蛋白质及其交互)的模型如 AlphaFold3 要么缺乏自然语言能力,要么依赖 adapter-based 设计,导致输入输出不对称——生物分子内容作为编码器特征输入,但模型无法原生生成非文本模态。这使得跨模态任务(如文本条件下的分子生成)和跨实体任务(如分子-蛋白质亲和力预测)需要专门的架构或融合模块,难以在单一框架下统一处理。
本文的目标是构建首个在单一 decoder-only 架构中同时实现原生多模态和广泛实体覆盖的生物基础模型,支持分子和蛋白质的序列、结构和自然语言的统一输入输出,无需外部编码器、投影适配器或模态特定输出头,在 80 个跨 6 个类别的下游任务上达到或超过专门模型的性能。
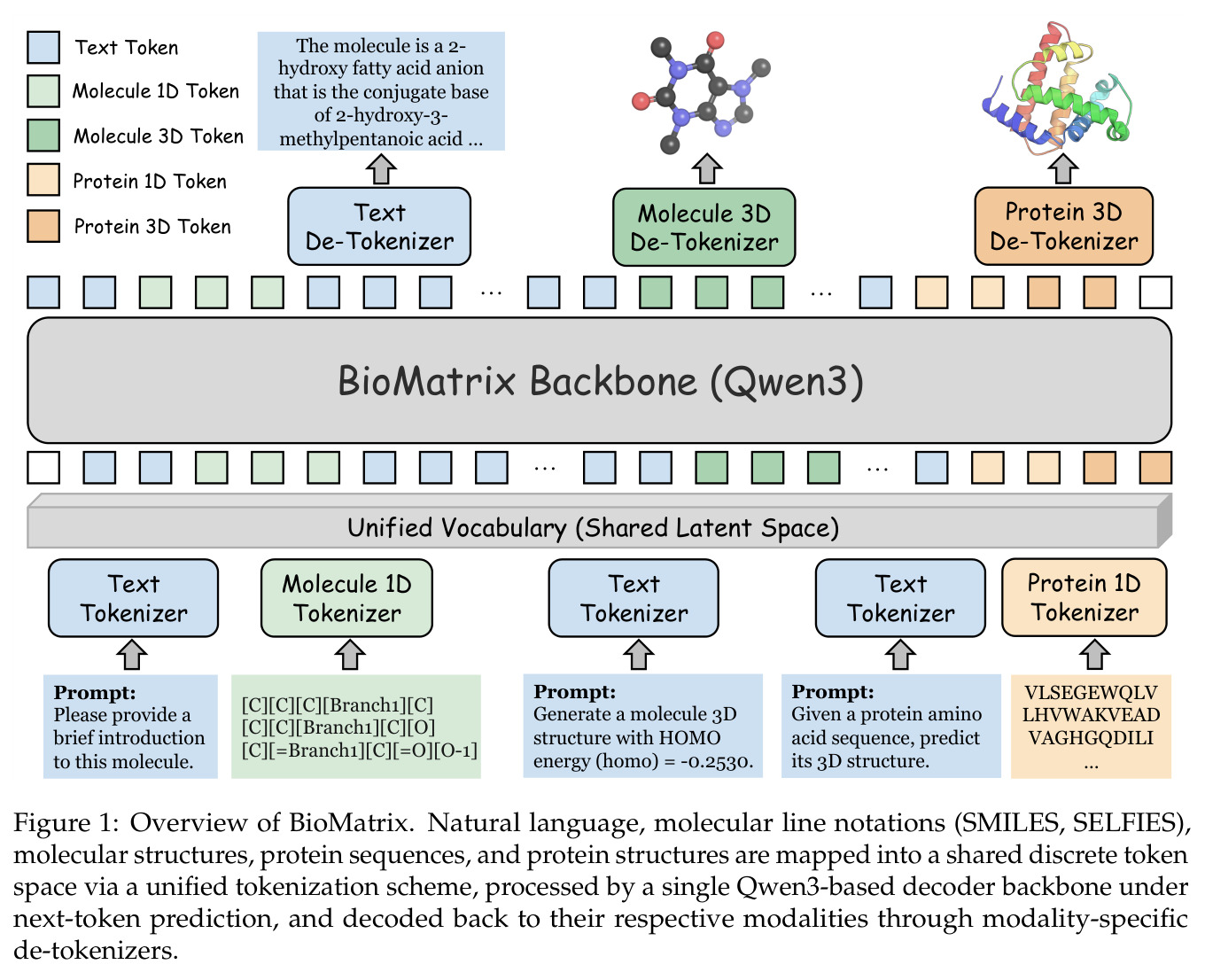
与已有工作不同的是,本文的切入点是将异构的 tokenizer 统一到单一离散 token 空间,而不是通过 adapter 融合多模态。具体而言,论文将分子序列(SMILES/SELFIES)、分子结构、蛋白质序列(氨基酸 token)、蛋白质结构和自然语言全部映射到扩展的 Qwen3 词汇表中,使用模态特定的边界 token 进行分割。这种设计使得本需不同架构的任务——如 captioning、文本条件设计、折叠、反向折叠、结构生成和交互预测——都变成了同一个 next-token 预测目标下的不同条件生成模式,彻底统一了技术框架。
核心方法
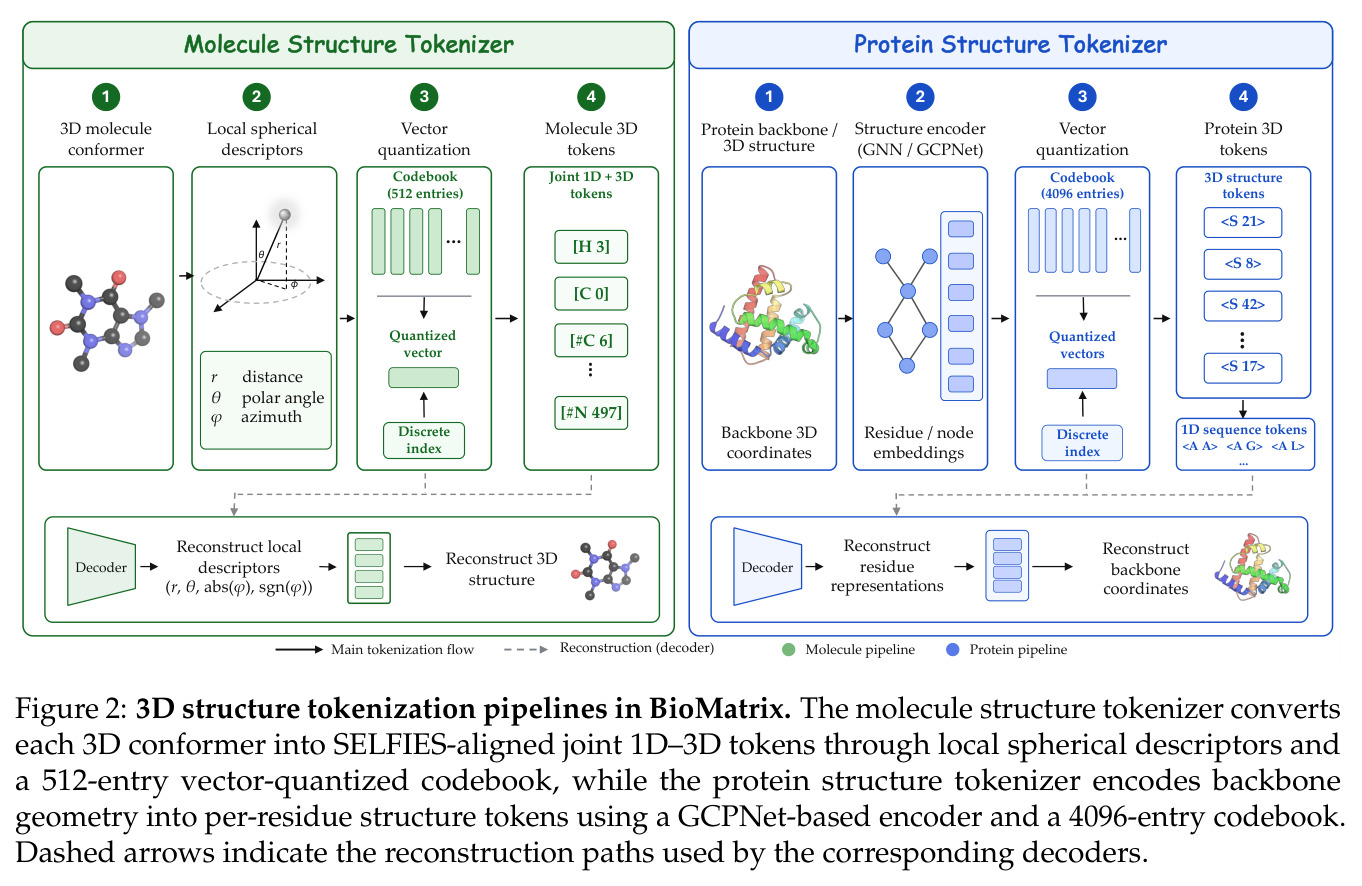
BioMatrix 的核心思想是将所有生物分子表示(文本、序列、结构)翻译成统一的离散 token 语言,然后用一个 decoder-only 语言模型来说这种语言。直觉上,就像一个翻译官可以同时用中文、英文和手语交流,不需要为每种语言准备专门的解码器。技术路线上,论文设计了统一的 tokenization 方案:文本和序列直接 token 化,连续的 3D 结构通过 SE(3)-invariant 的 VQ tokenizer 离散化,然后用模态边界 token 标记不同模态的段落,在单个 next-token 预测目标下进行训练。分子结构 tokenizer 使用局部球坐标系描述每个原子相对于参考帧的位置,蛋白质结构 tokenizer 按残基粒度编码骨架几何。
核心创新点是将异构的模态 tokenizer 统一到单一离散 token 空间,使得所有任务都变成 next-token 预测,而不是通过 adapter 融合多模态。与已有工作相比,BioMatrix 不需要专门的编码器来处理生物分子,不需要投影层来对齐不同模态的表示空间,也不需要为每种输出模态准备不同的解码头。所有输入和输出都在同一个 token 流中,只是用特殊的边界 token 标记不同模态的段落。例如,分子-蛋白质交互预测只需要将分子的 SELFIES token 和蛋白质的残基 token 并排放在同一个序列中处理,不需要专门的几何交叉注意力机制。这种设计的关键是 residue-level 对齐的蛋白质 tokenizer(每个氨基酸对应一个序列 token 和一个结构 token)和分子 SELFIES-token 对齐的结构 tokenizer(每个原子对应一个联合 token)。
方法步骤详情
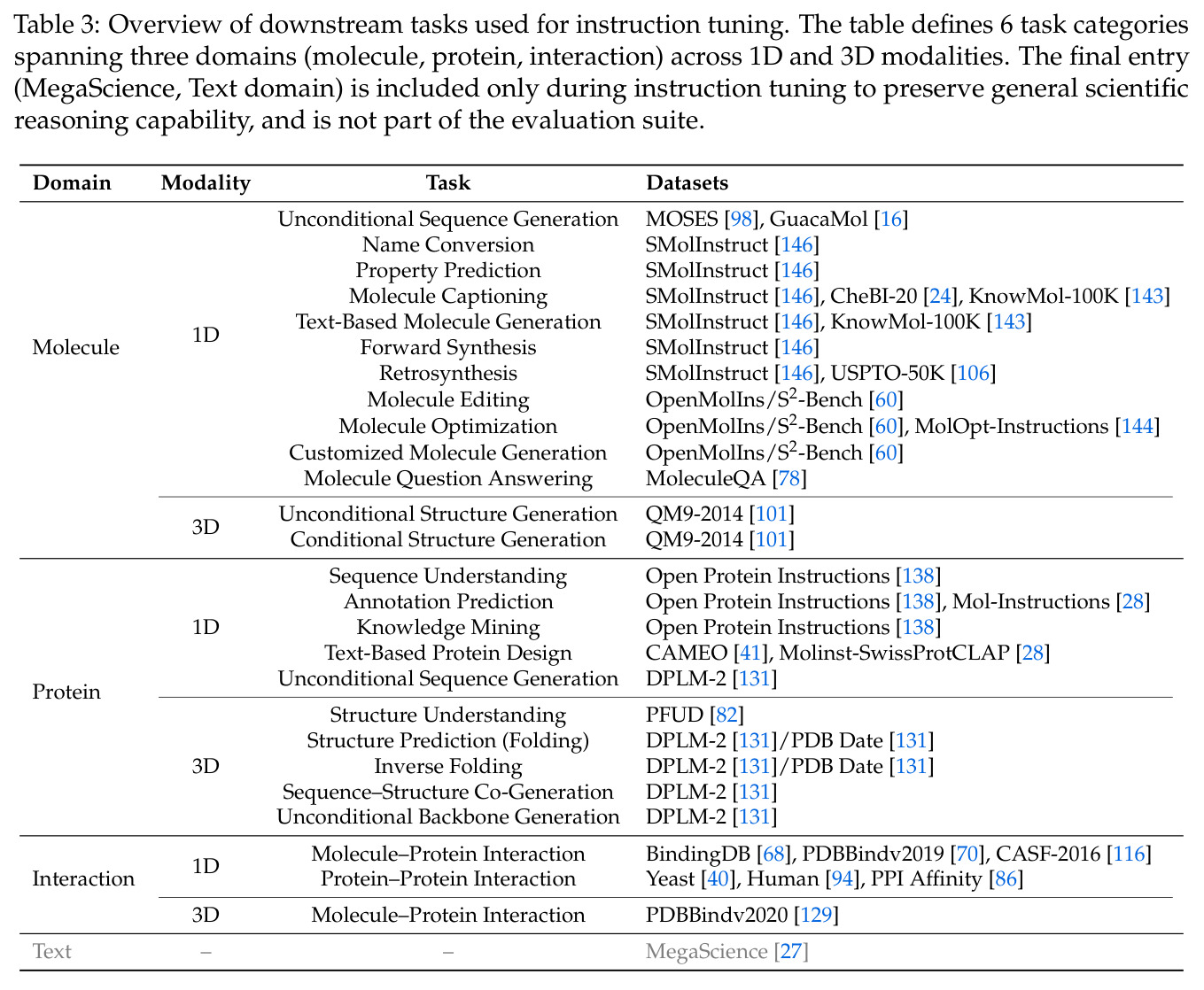
方法分为四个主要步骤:首先,统一 tokenization 方案。对于文本和序列,直接使用 Qwen3 tokenizer(SMILES)或添加新 token(SELFIES、氨基酸);对于 3D 结构,使用专门的 VQ tokenizer——分子使用基于局部球坐标系的 MolStrucTok,将每个原子的位置表示为包含径向距离、极角、方位角绝对值和方位角符号的四维向量,然后量化到 512 条目码本;蛋白质使用基于 GCPNet 的 GCP-VQVAE,将每个残基的骨架几何量化到 4096 条目码本。第二,扩展词汇表。添加模态特定的 token(分子 1D/3D、蛋白质 1D/3D)和结构控制 token,通过基于描述的初始化方案用 Qwen3 的预训练嵌入矩阵初始化新 token。第三,持续预训练。在 3044 亿 token 的语料库上训练,包括通用文本、科学文本、分子数据、蛋白质数据和跨实体数据,训练使用随机组合策略打乱字段顺序,对蛋白质采用三视图模式。第四,指令微调。在 80 个任务上分组微调,分为分子 1D、分子 3D、蛋白质 1D 理解、蛋白质 1D 设计、交互等组别,每个任务使用多样化的 prompt 模板。
技术新颖性
BioMatrix 的技术新颖性主要体现在三个层面:一是架构层面的统一性,首次在单一 decoder-only 模型中原生支持分子和蛋白质的序列、结构和语言,避免了 adapter-based 方法的输入输出不对称问题;二是 tokenization 方面的创新,设计了 residue-level 对齐的蛋白质 tokenizer(保持 AA token 和结构 token 的一一对应)和分支解耦的分子结构 decoder(将异构的几何目标分离到不同的 MLP head,提高重建精度);三是训练策略层面的完整,构建了 3044 亿 token 的多模态持续预训练语料库,采用随机组合和三视图模式暴露模型到孤立和联合的模态视图。与 ESM3(仅蛋白质、无文本)和 BioMedGPT(adapter-based)相比,BioMatrix 的统一 token 空间使得跨模态和跨实体任务不需要额外的融合模块。
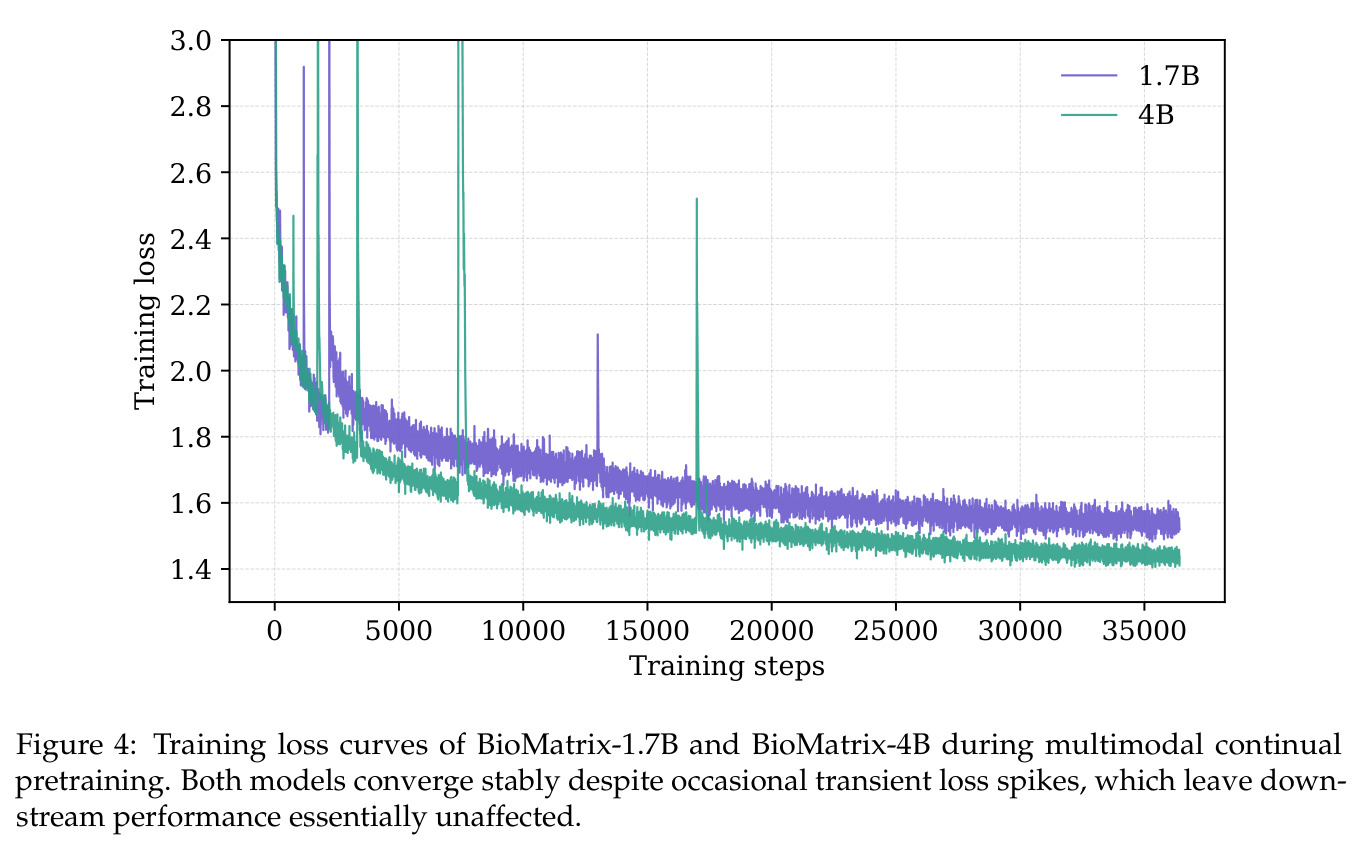
实验结果
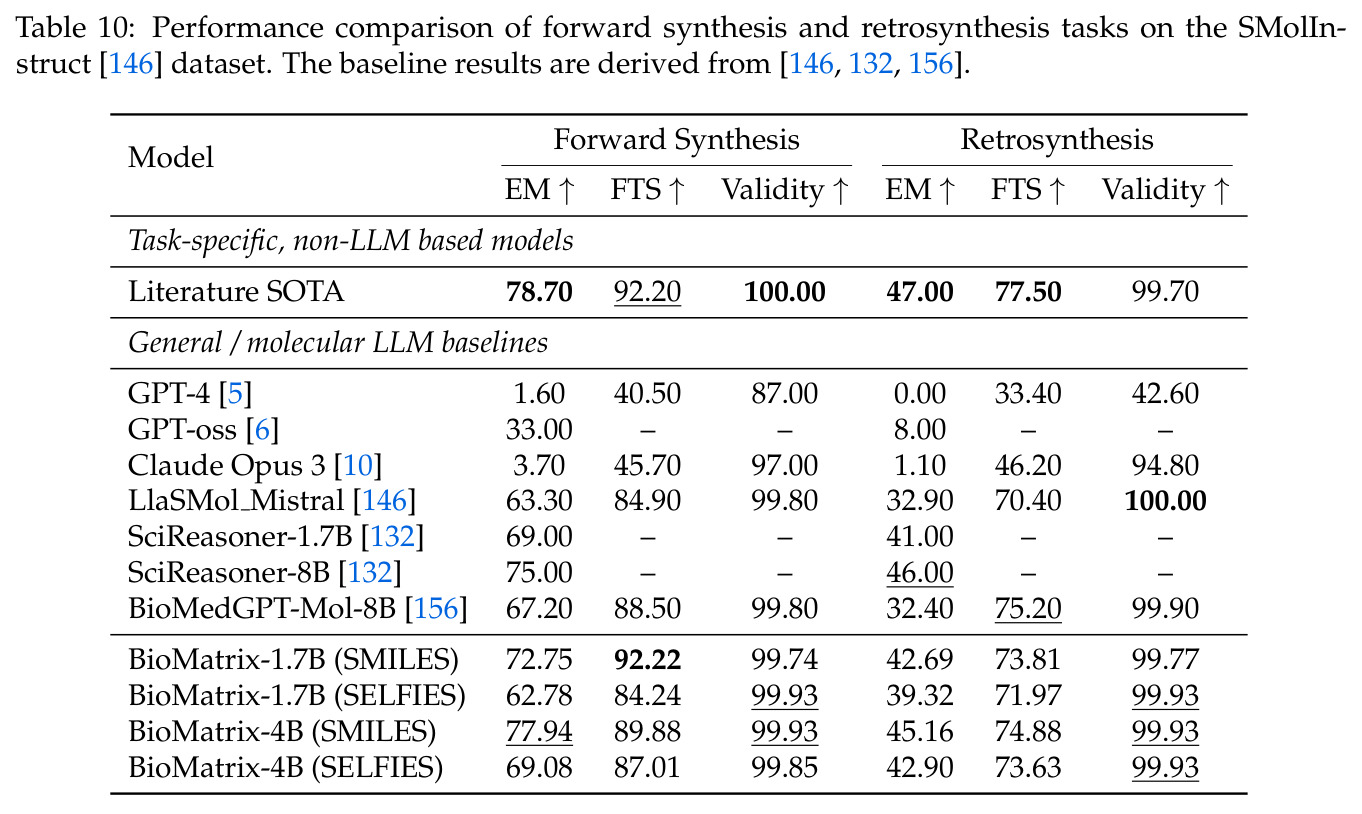
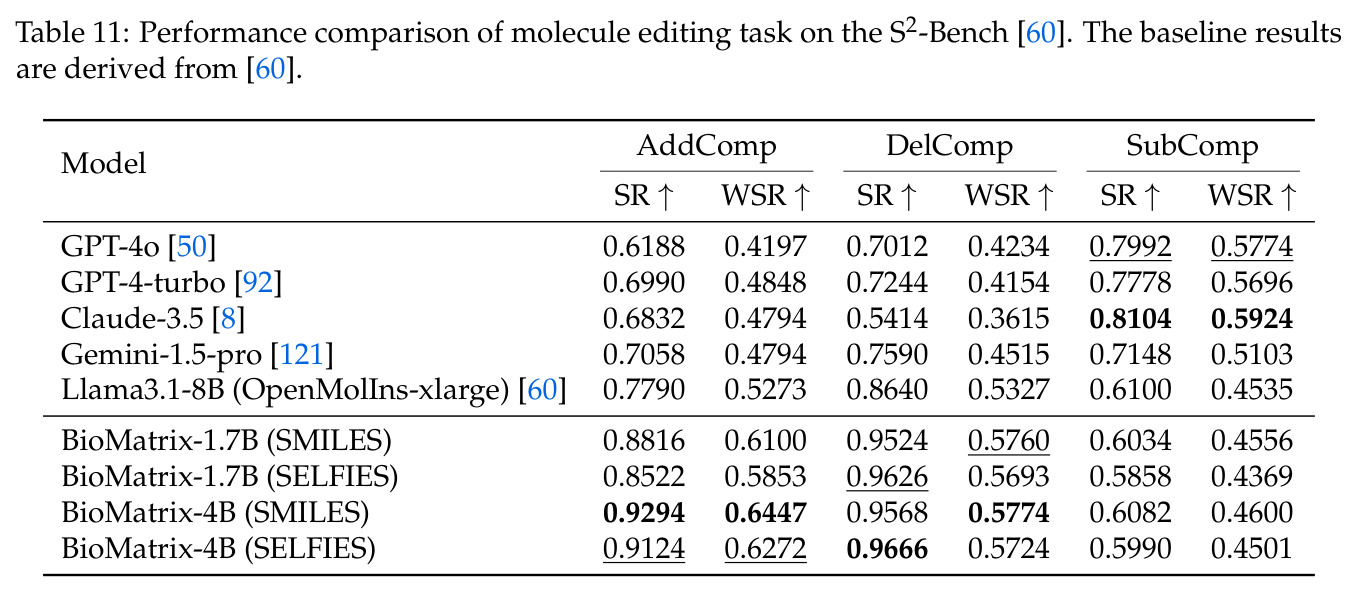
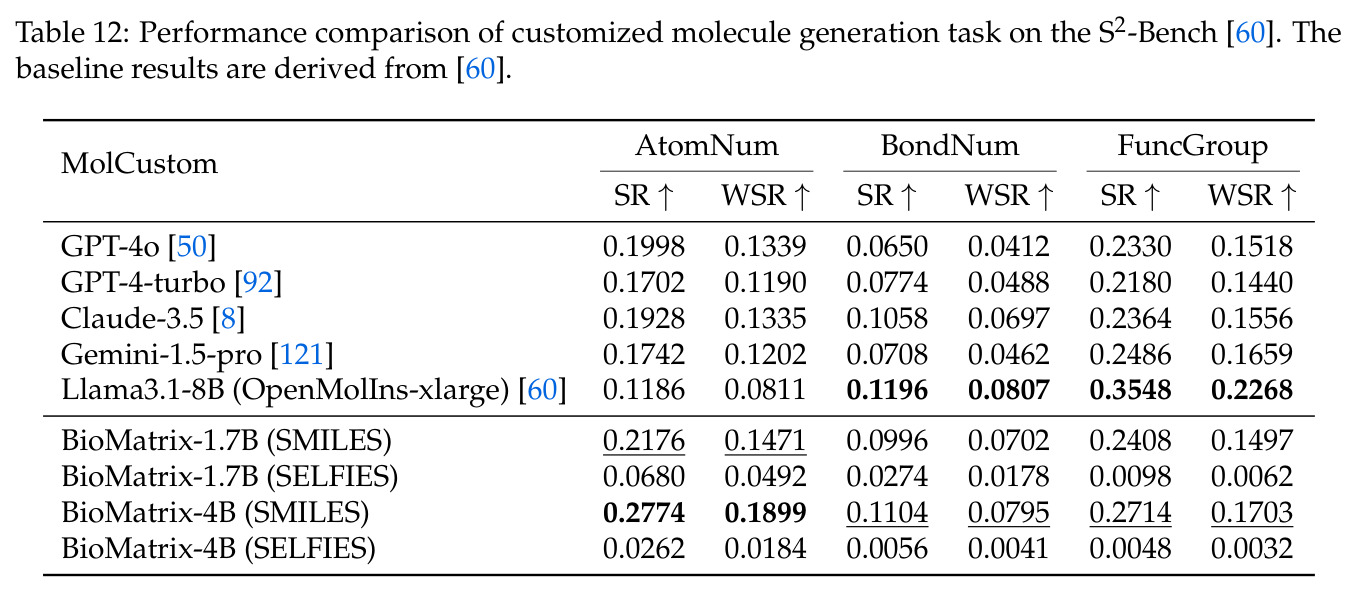
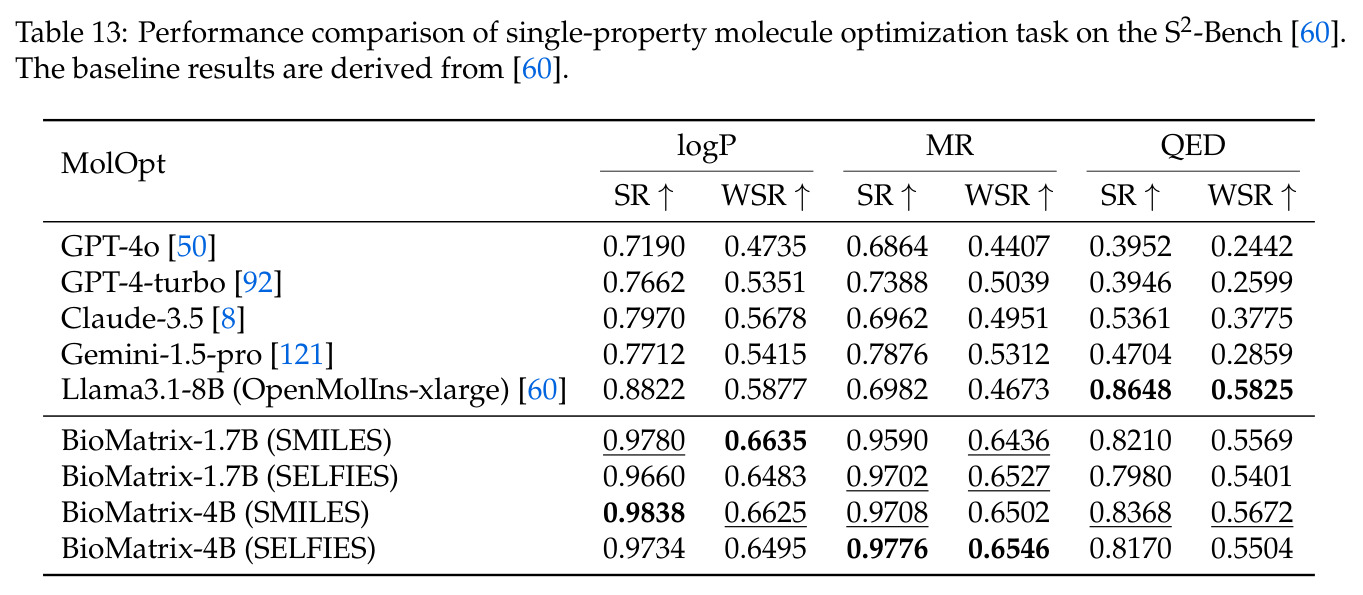
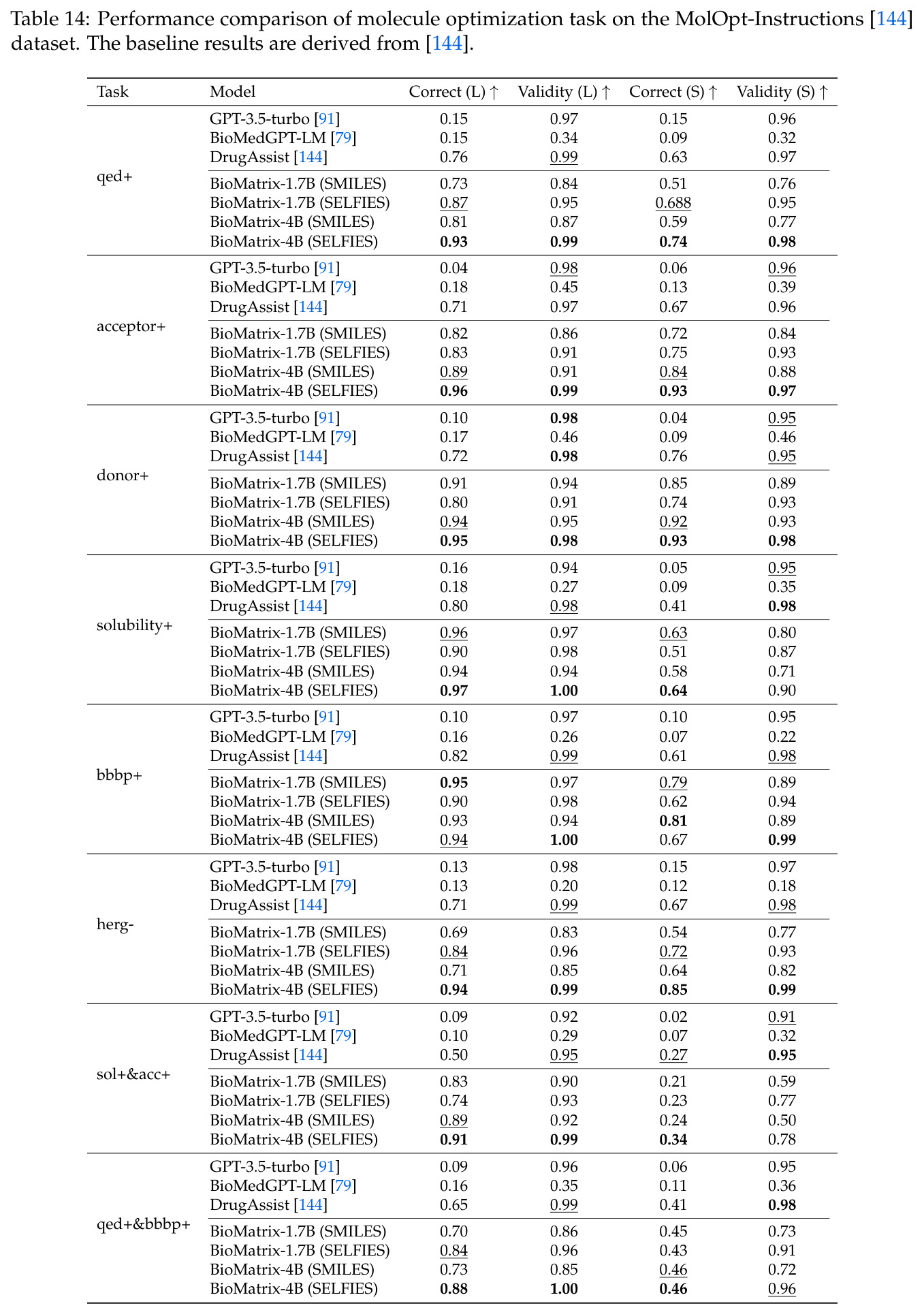
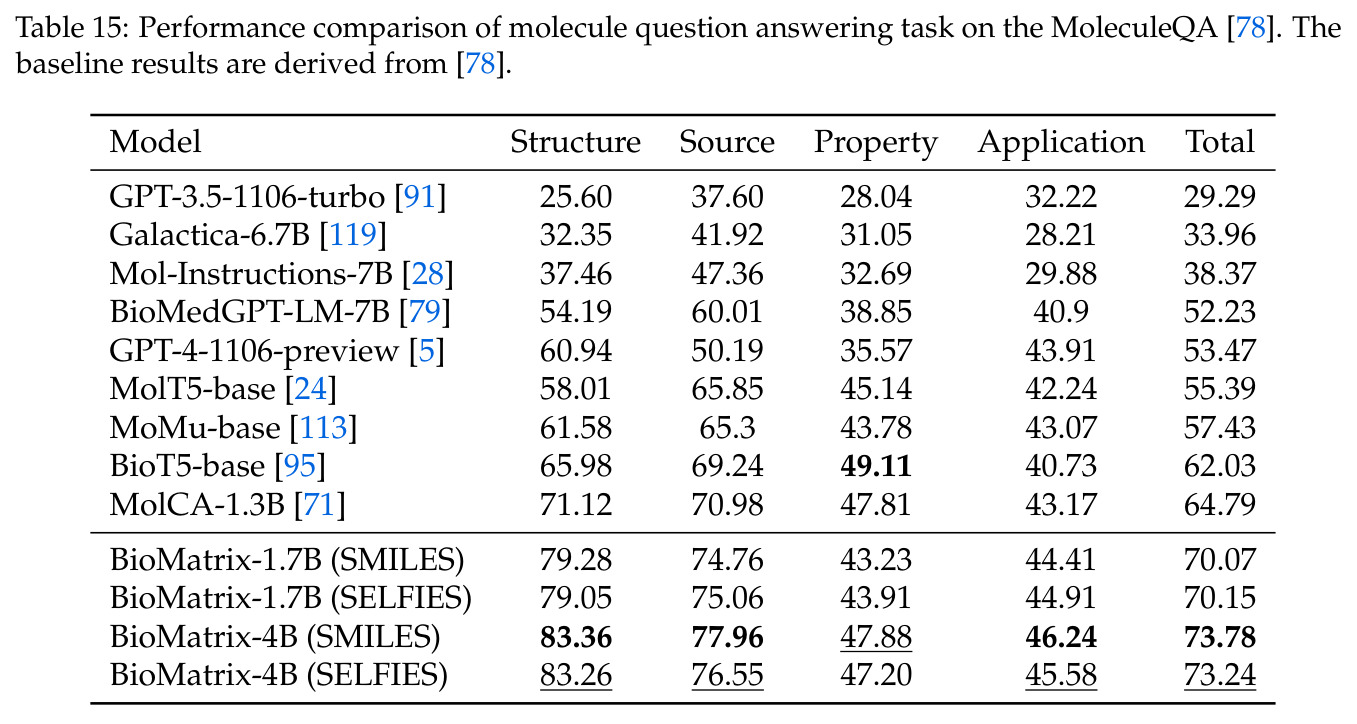
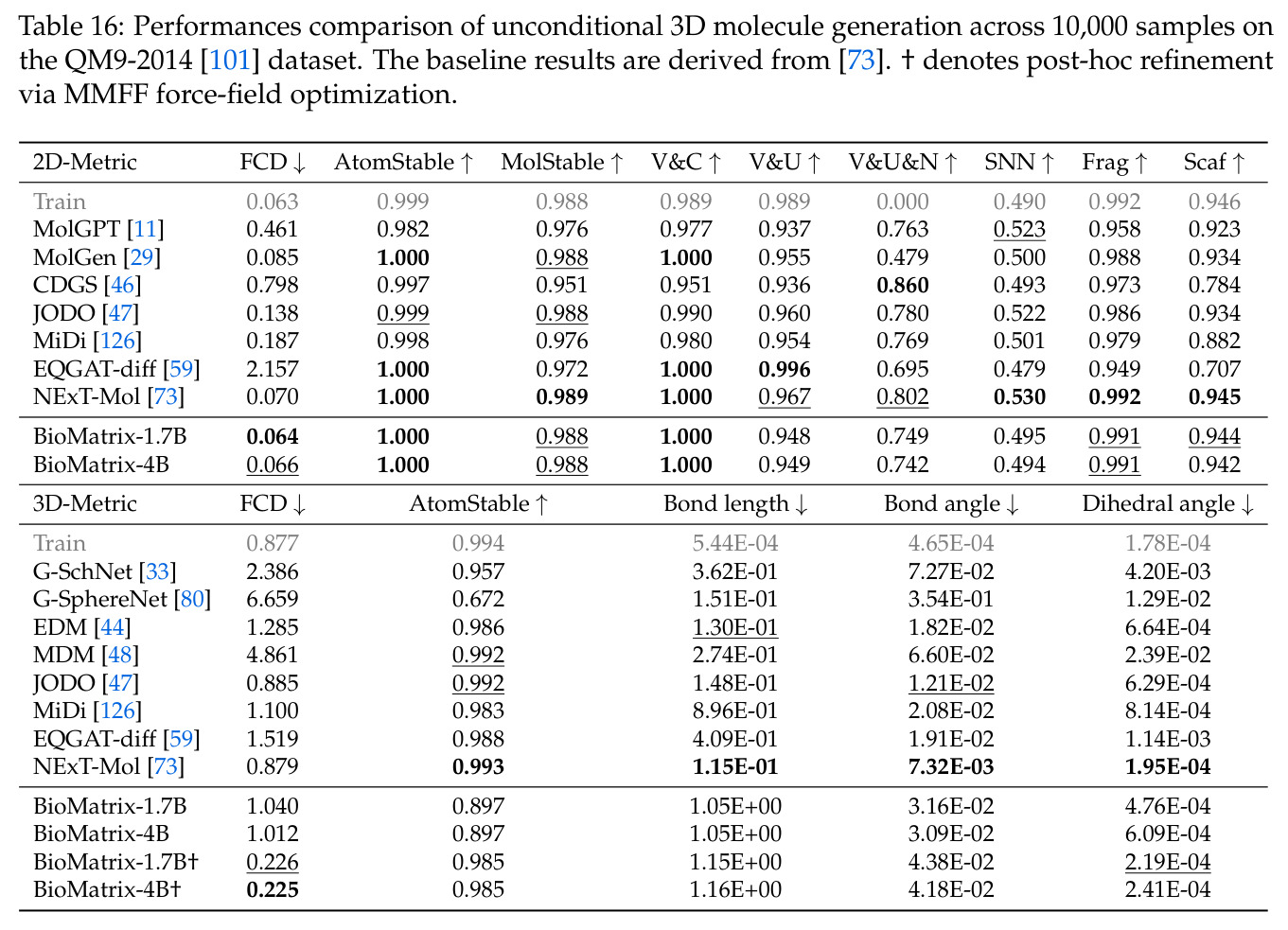
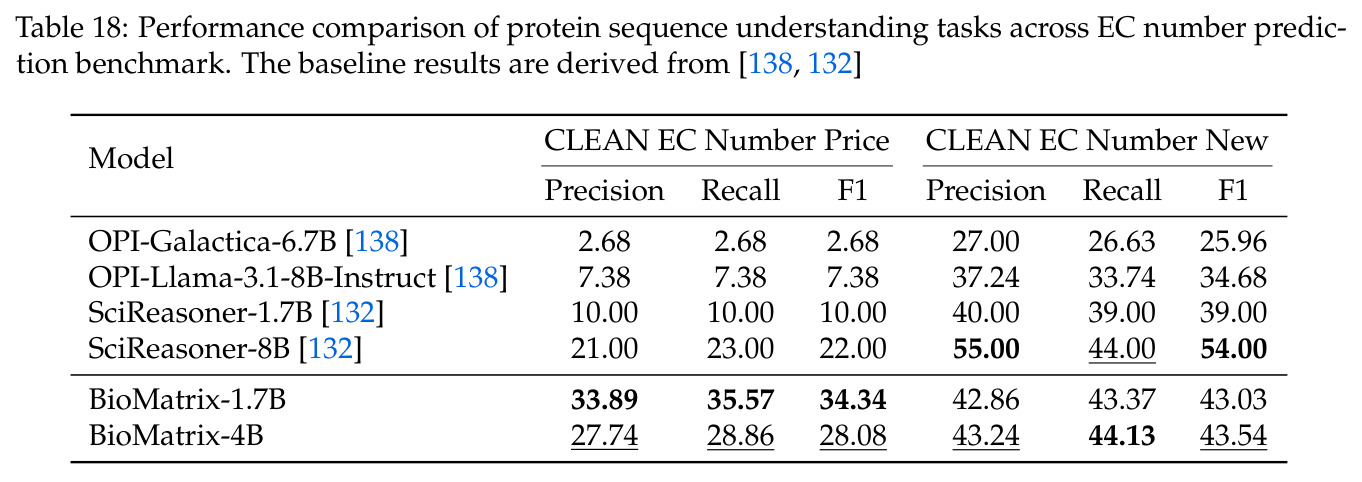
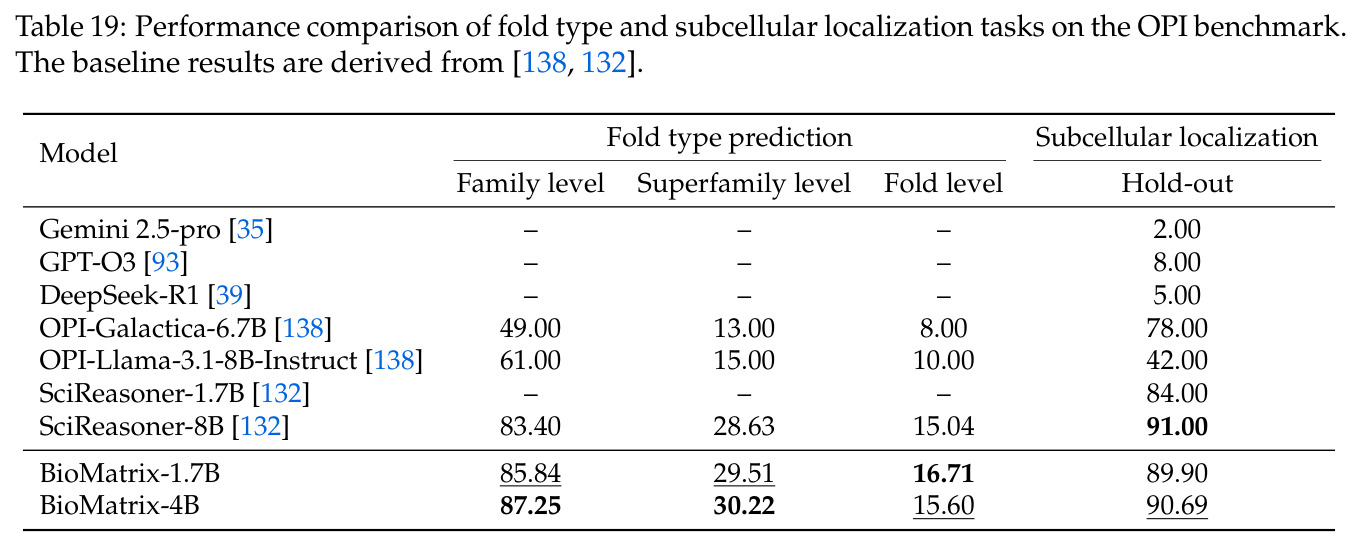
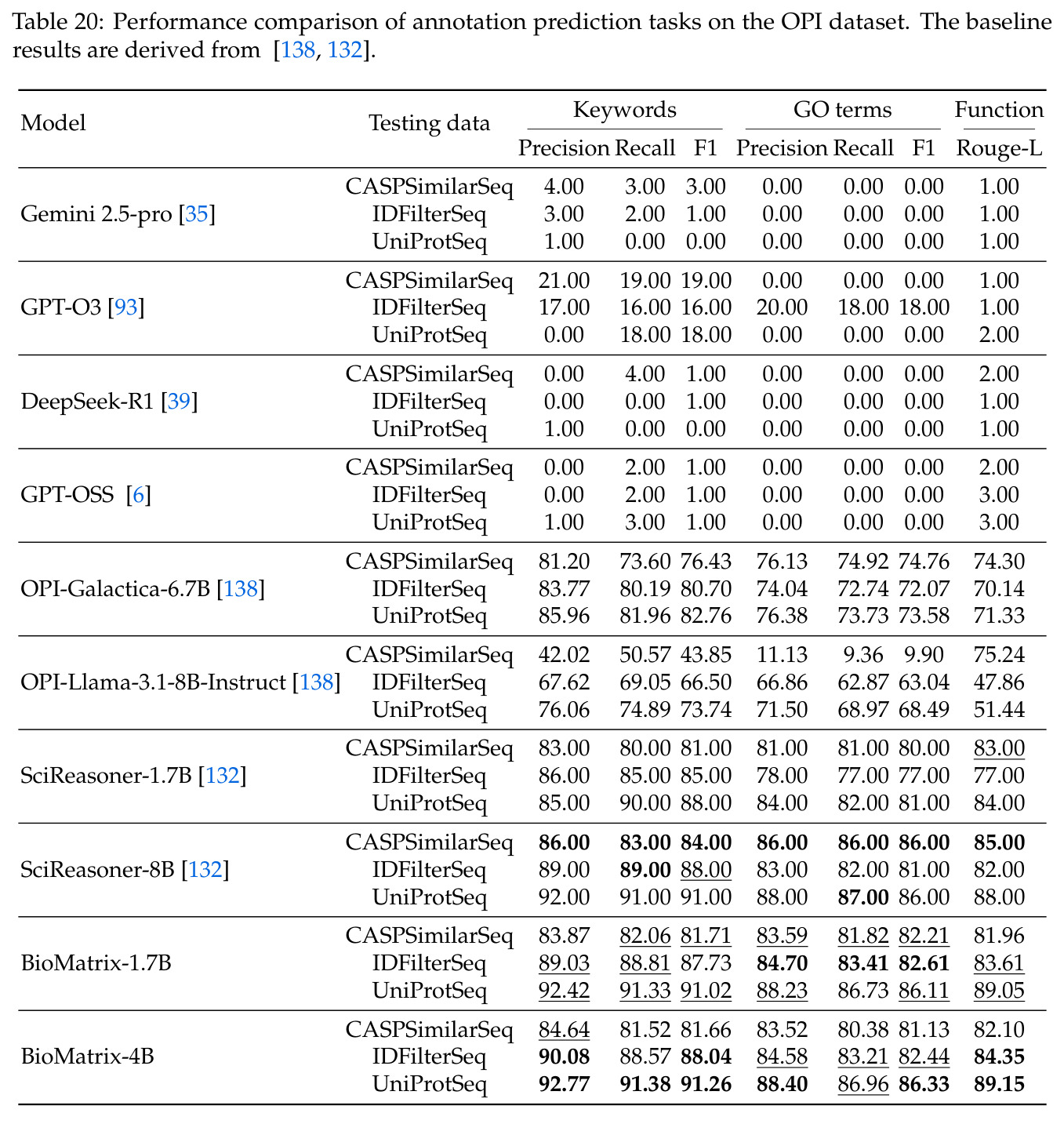
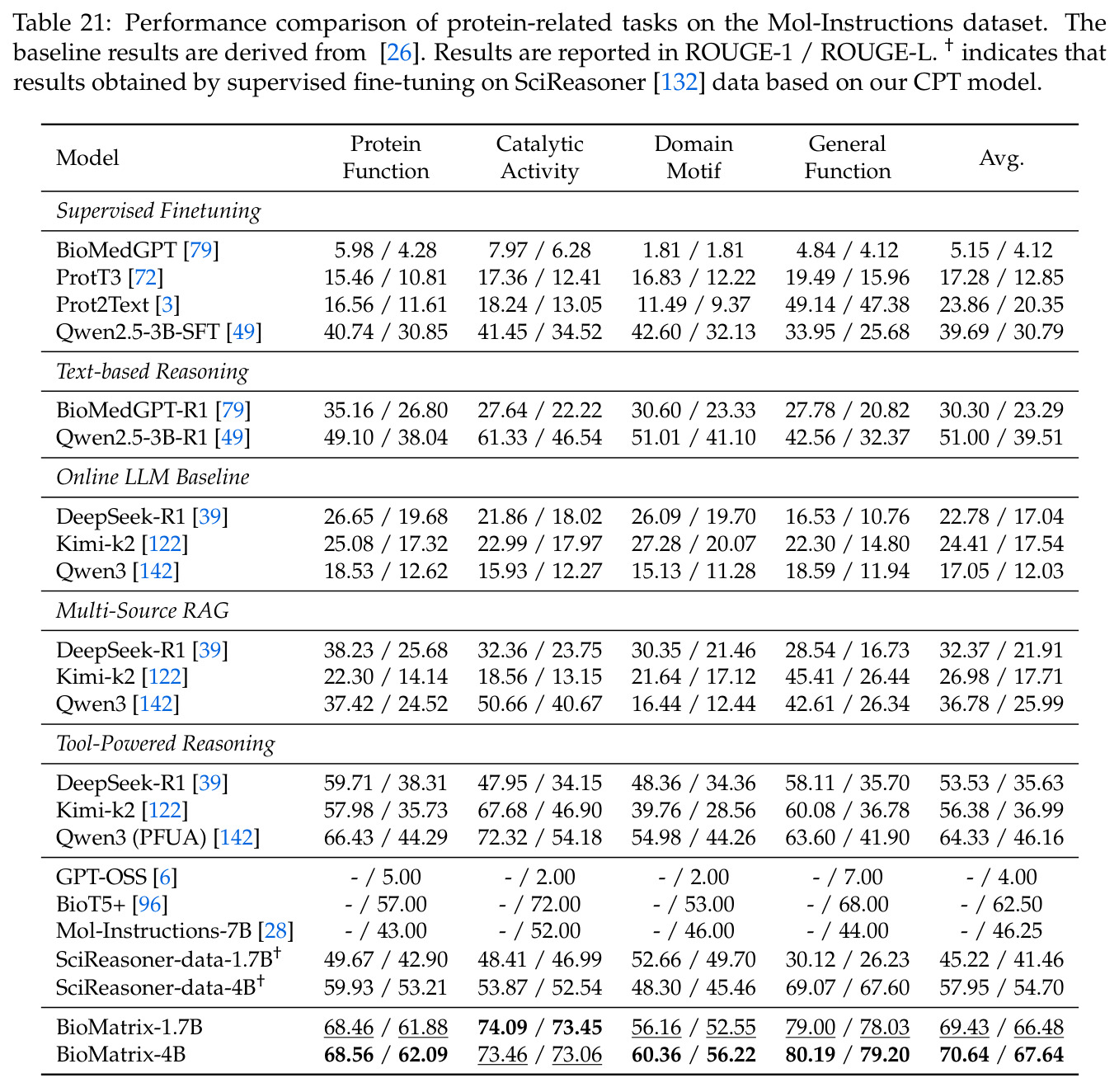
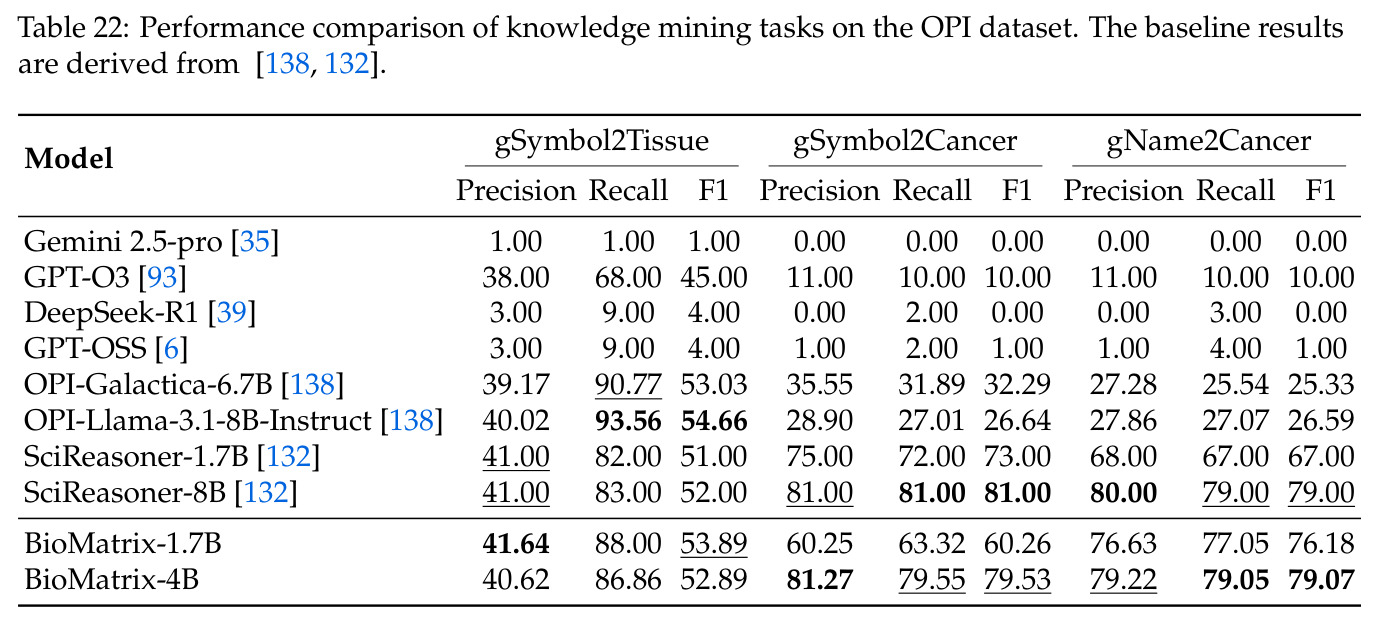
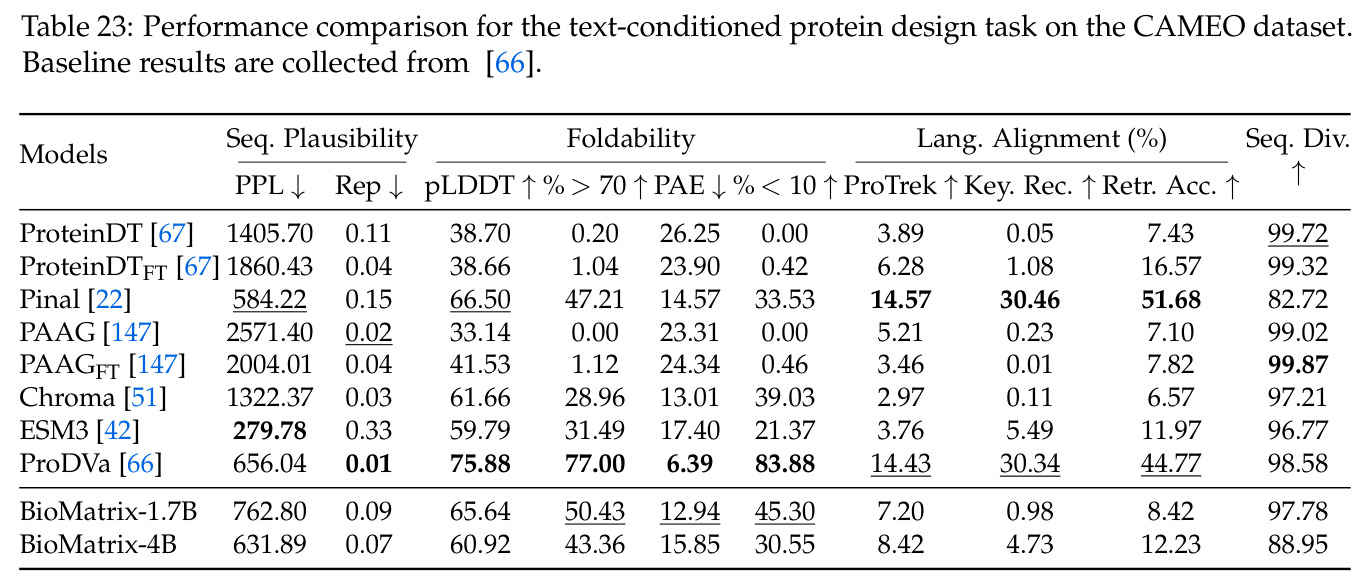
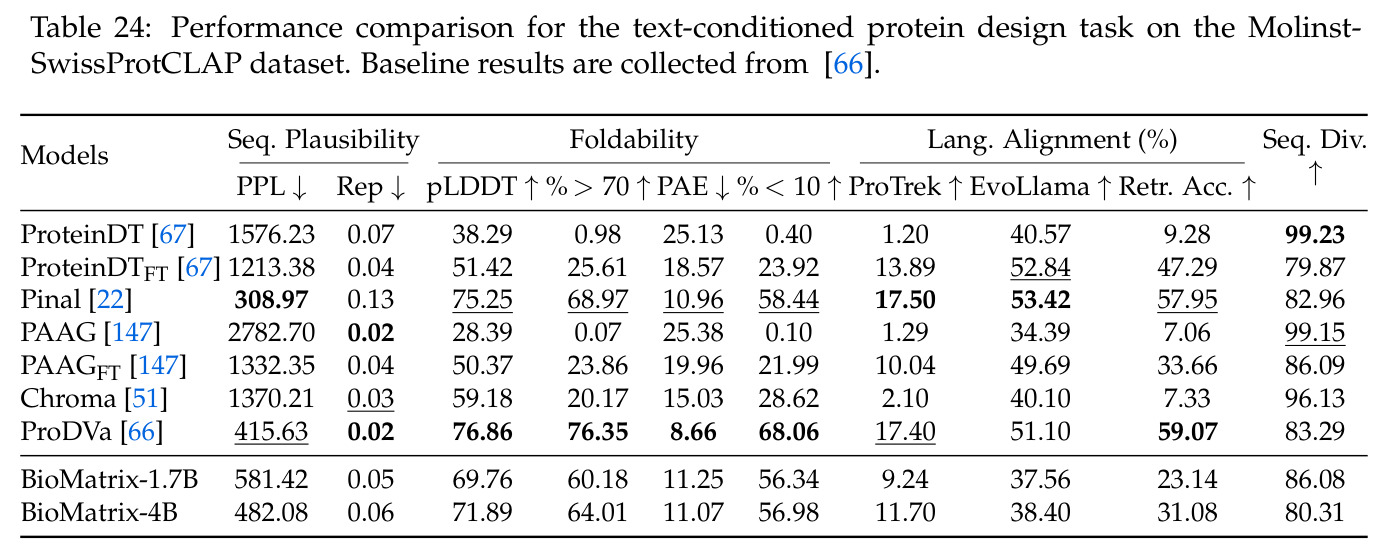
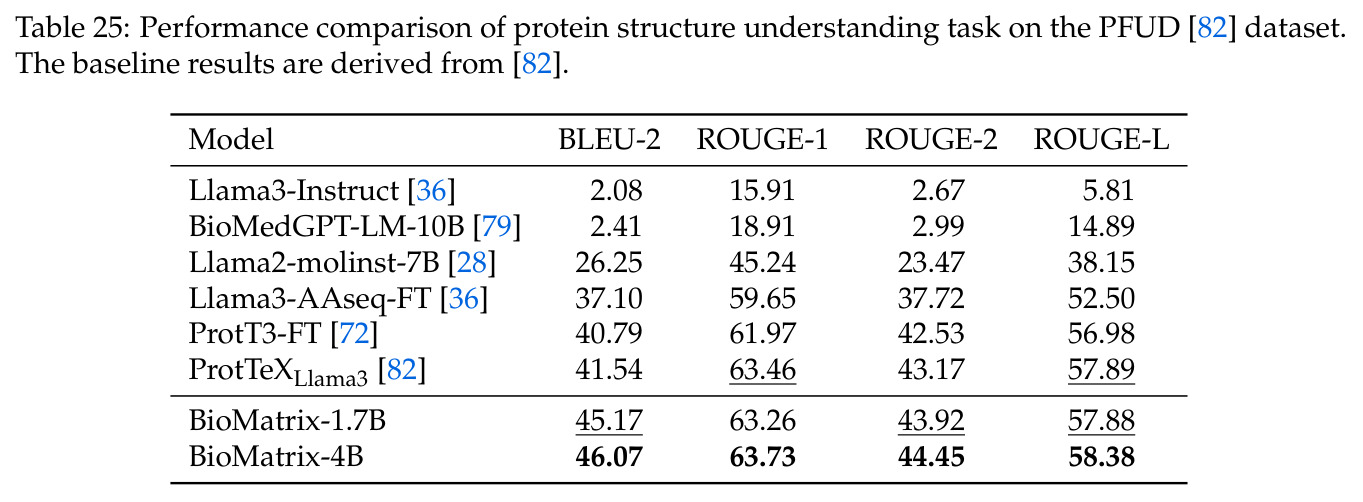
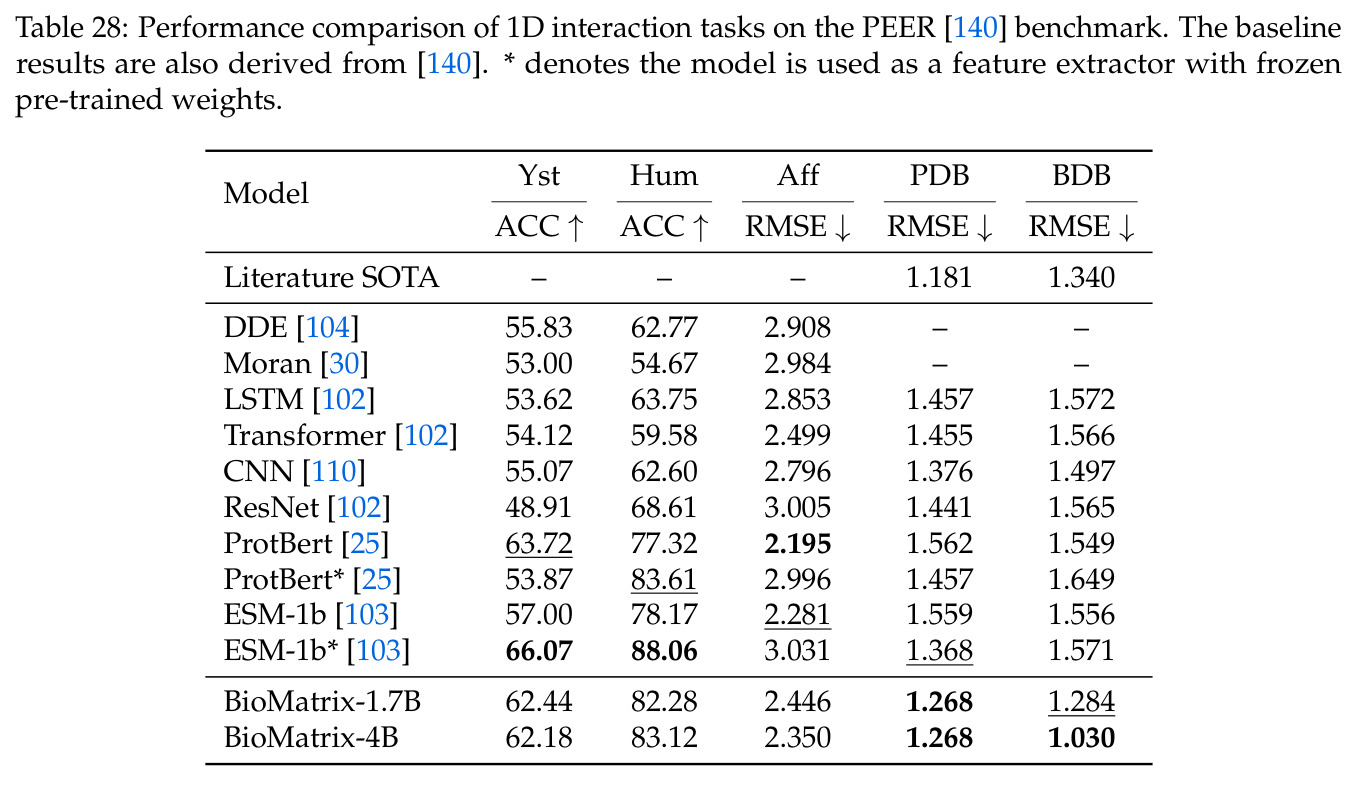
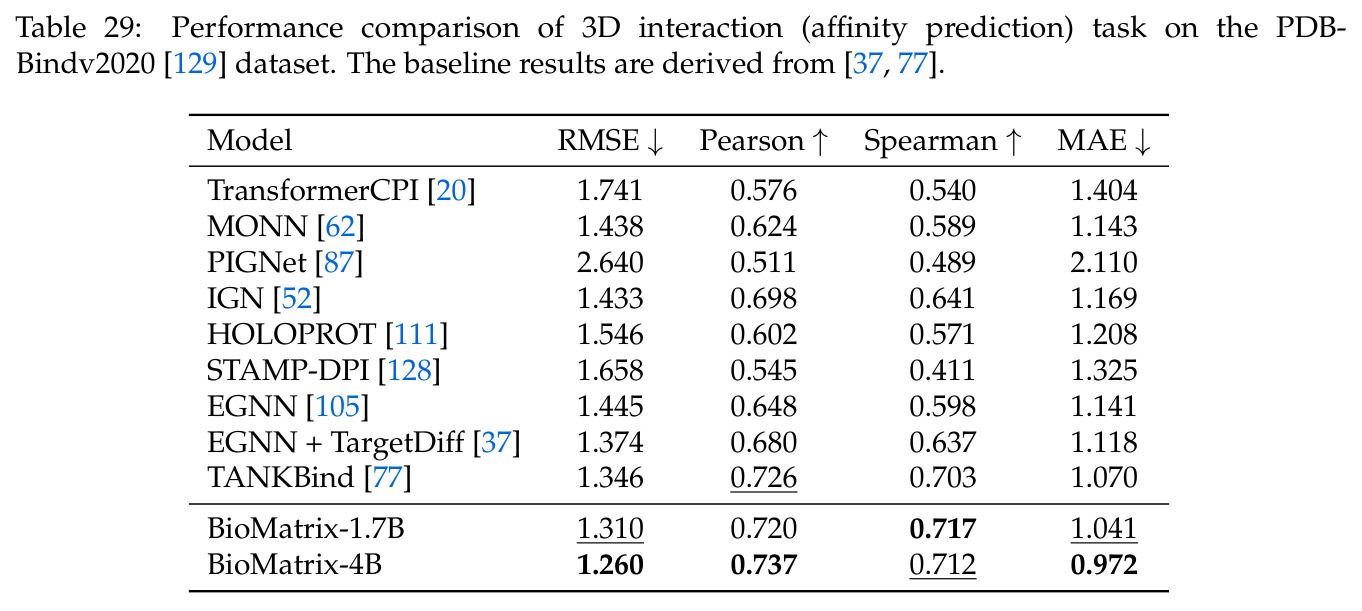
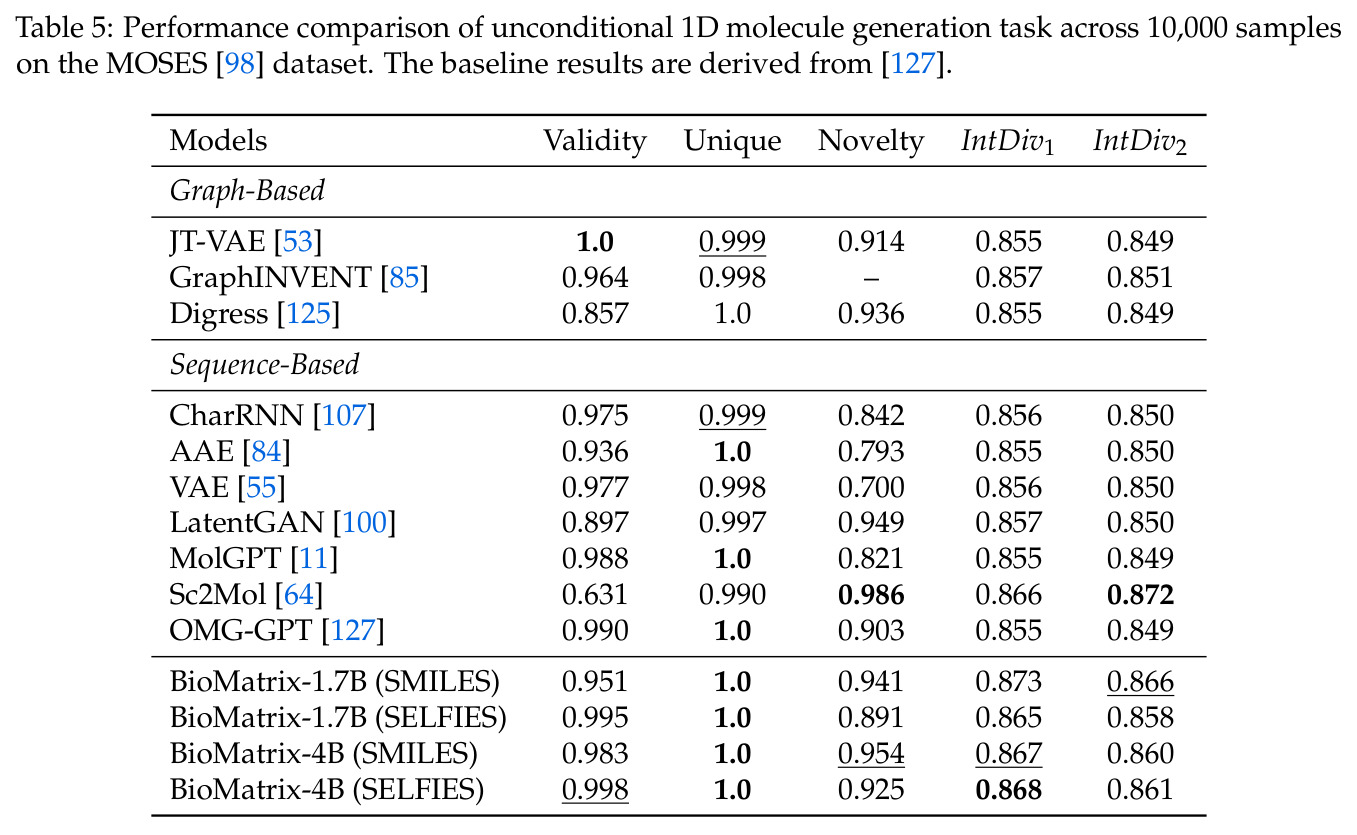
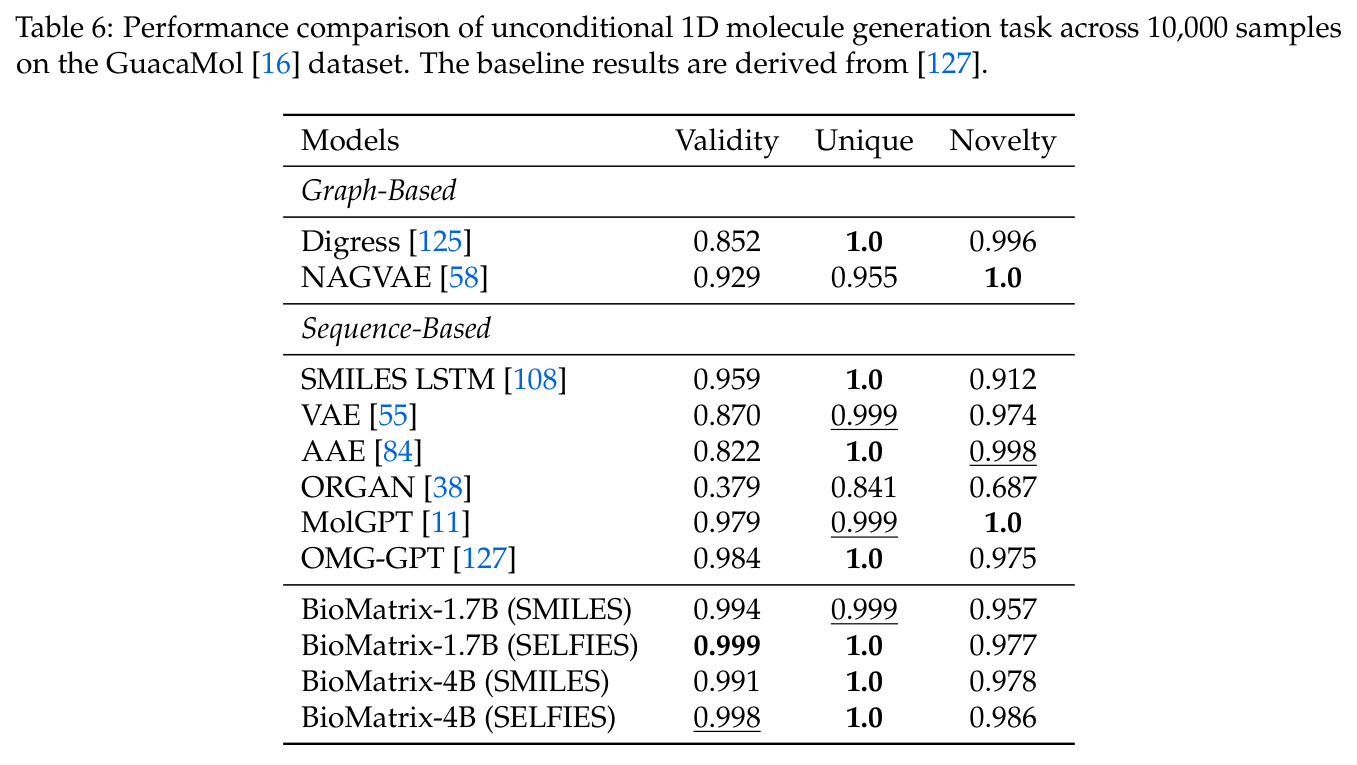
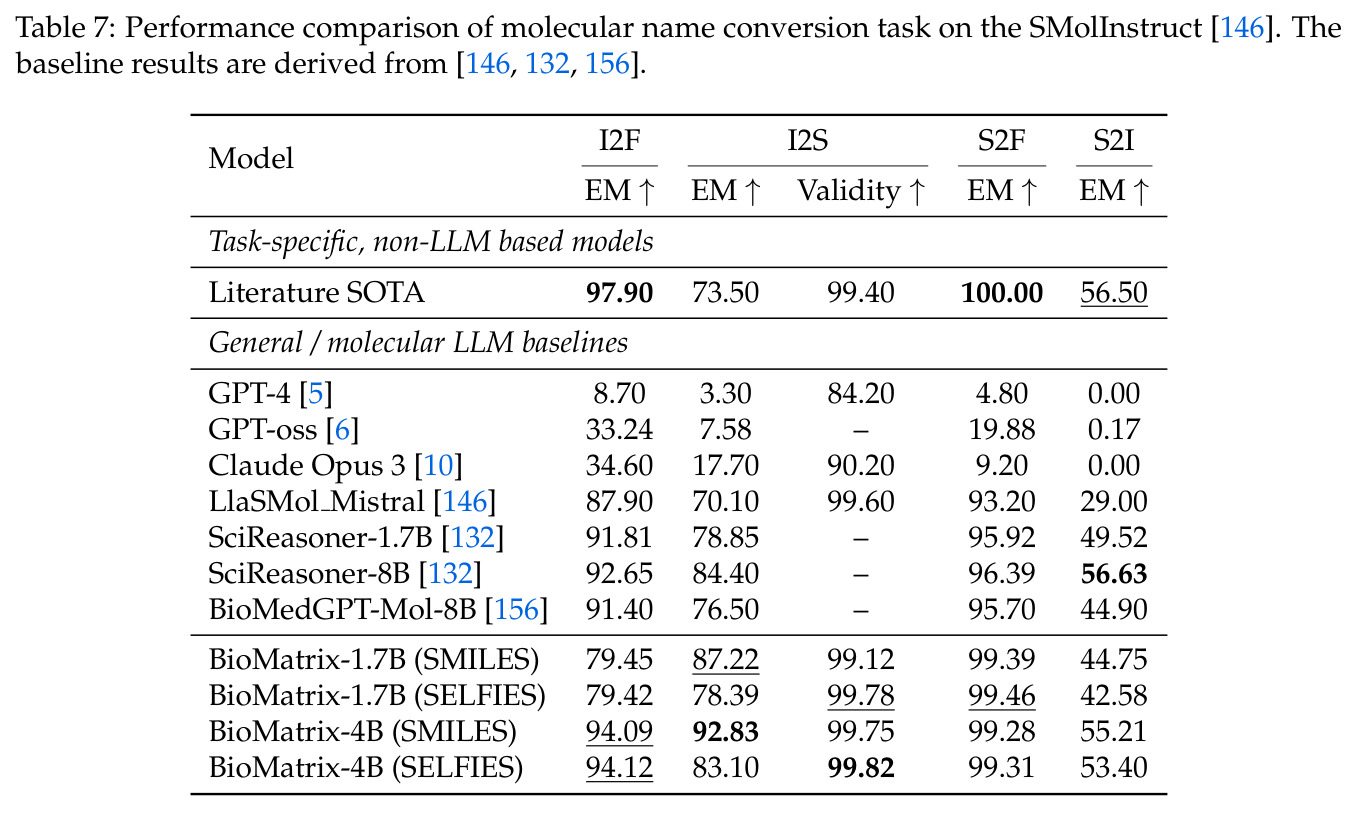
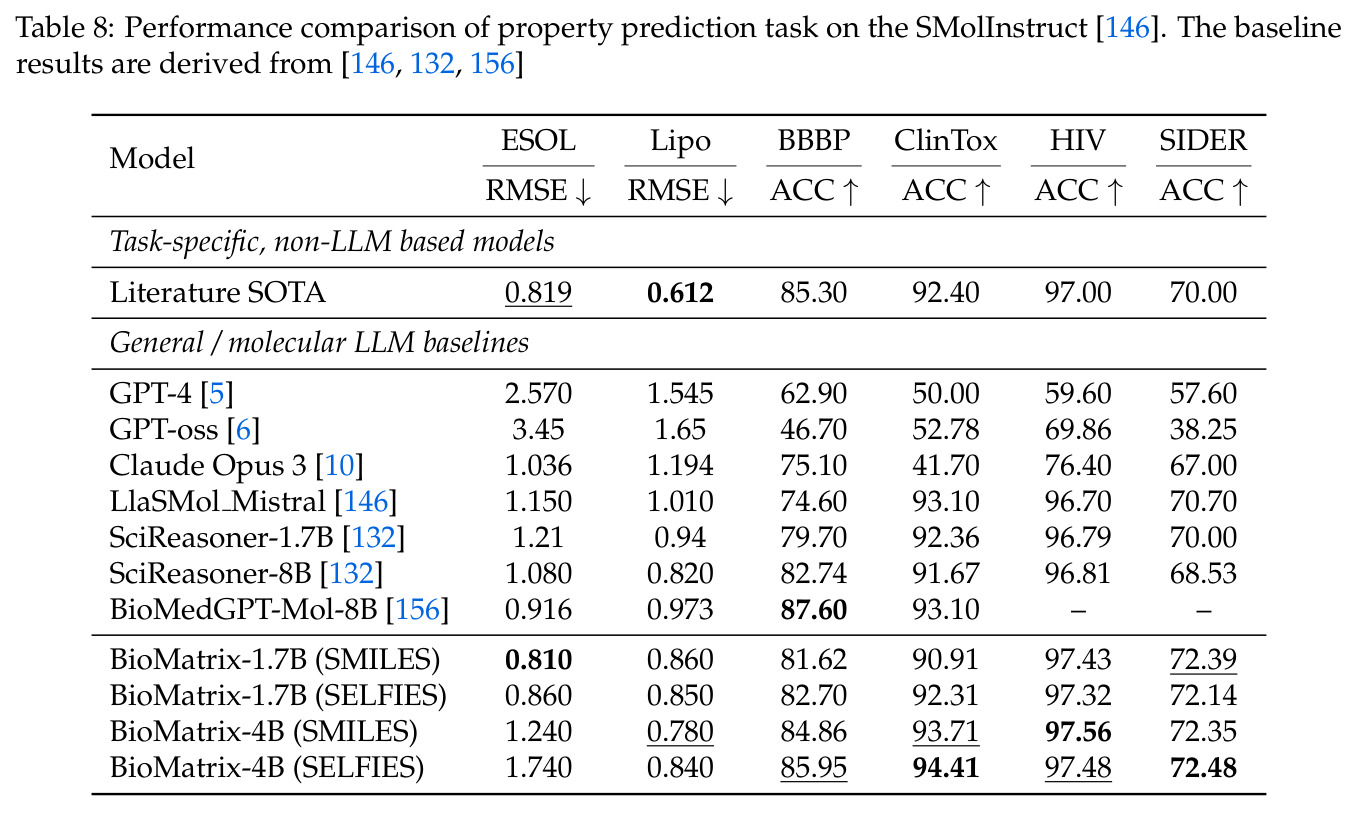
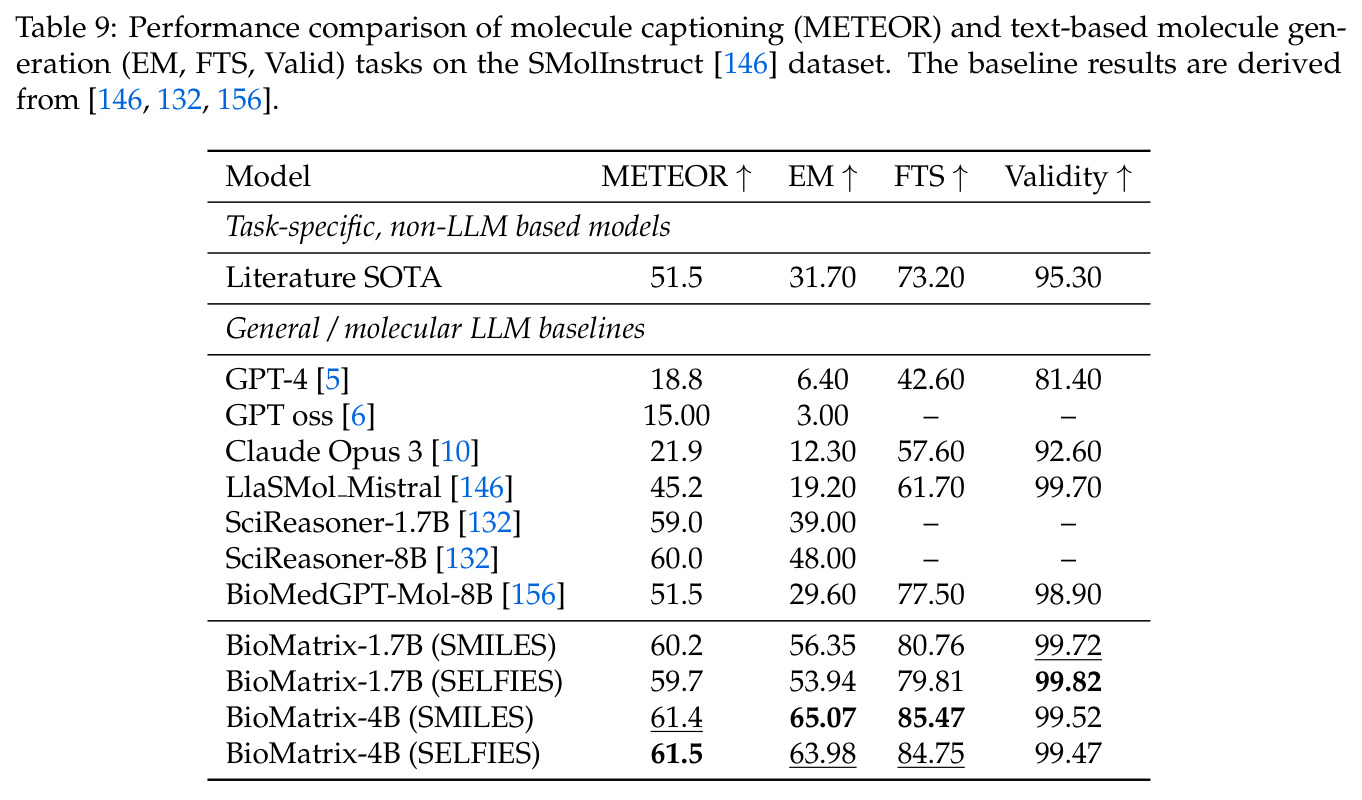
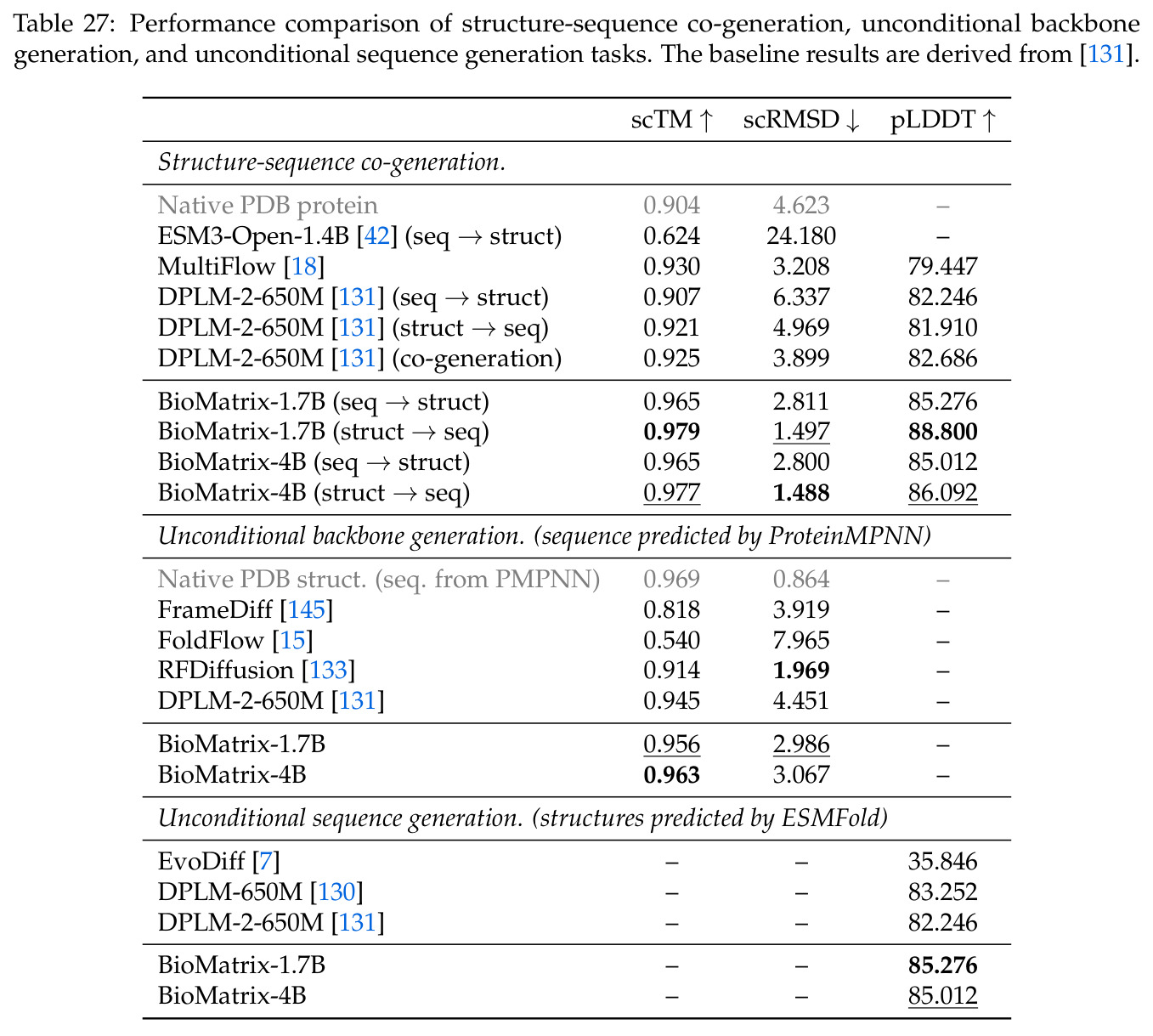
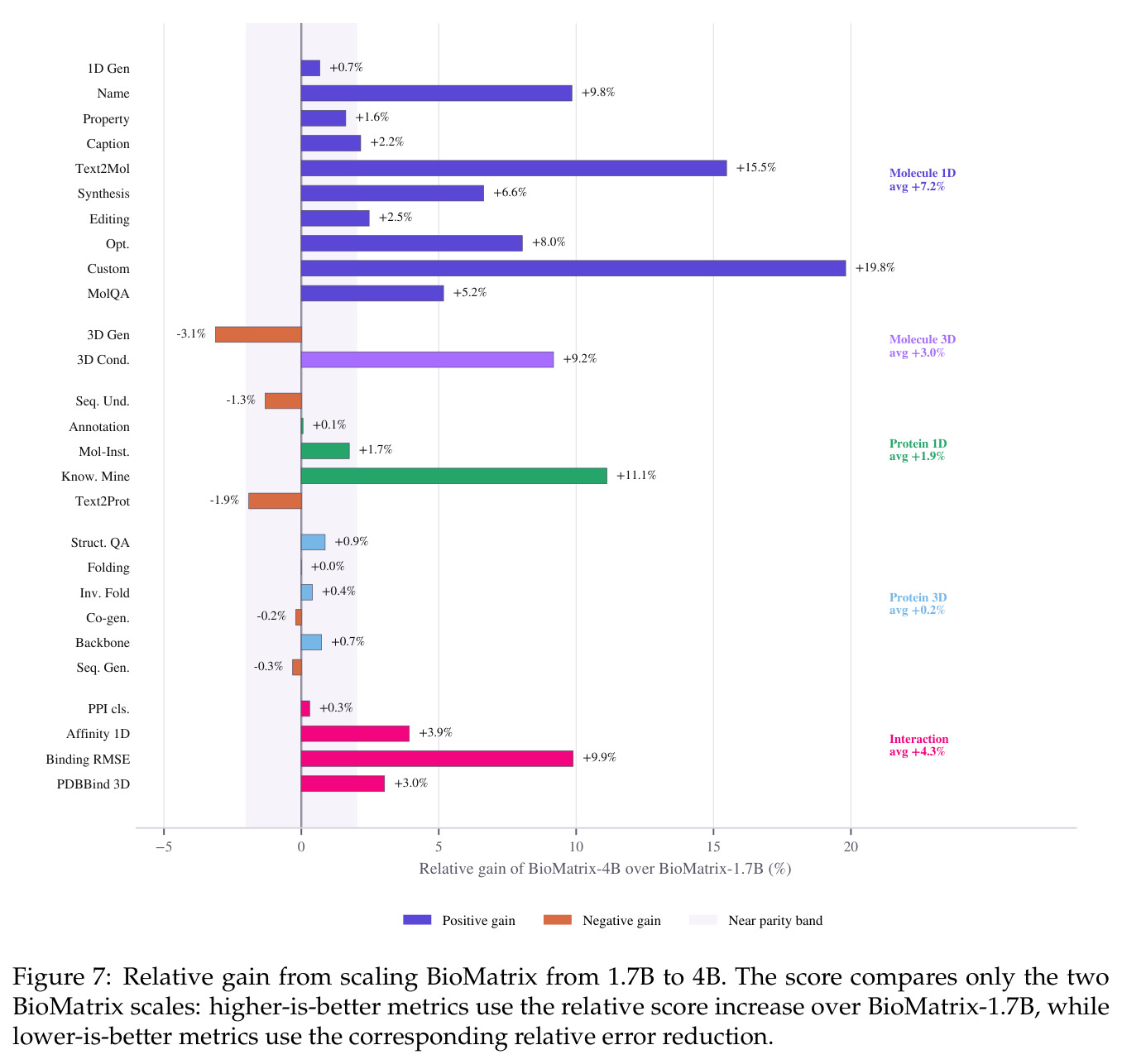
BioMatrix 在 80 个任务中的 77 个上达到 SOTA 或竞争性能,验证了统一多模态框架的可行性。分子任务方面,无条件生成在 MOSES 和 GuacaMol 上达到接近饱和的有效性(0.951-0.999)和唯一性(约 1.0),SELFIES 变体在化学有效性上占优;文本基分子生成在 METEOR(61.5)和 EM(65.07%)上超越 SciReasoner-8B(60.0/48.00%);分子编辑在 AddComp 上达到 SR 0.9294,超越 Llama3.1-8B(0.7790);属性条件 3D 生成在 QM9-2014 上将电子结构目标的 MAE 相比 NExT-Mol 降低 3-4 倍(HOMO:205→53 meV,LUMO:235→54 meV,Gap:297→81 meV)。蛋白质任务方面,序列理解在 EC Number Price 上达到 F1 34.34%,超越 SciReasoner-8B(22.00%);Fold type prediction 达到 Family 层 87.25%;反向折叠在 PDB date split 上达到 AAR 75.50%,超越 DPLM-2-3B(61.67%);序列-结构联合生成在 struct→seq 方向达到 scTM 0.979/scRMSD 1.50,超越 native-PDB 参考(0.904/4.62);无条件骨架生成达到 scTM 0.963,超越 RFDiffusion(0.914)。交互任务方面,分子-蛋白质亲和力预测在 PDBBindv2020 上达到 RMSE 1.260,超越 TankBind(1.346),且不需要专门的几何交叉注意力;BindingDB 上 RMSE 达到 1.030,超越文献 SOTA(1.340)。论文还发现了有趣的模式:SELFIES 在需要化学有效性的任务(无条件生成、属性优化)上占优,SMILES 在需要表面结构锚定的任务(定制化生成、合成)上占优;从 1.7B 到 4B 的缩放在知识密集型任务(分子命名转换、知识挖掘)上提升明显,但在已饱和的任务(无条件生成、反向折叠)上提升有限。
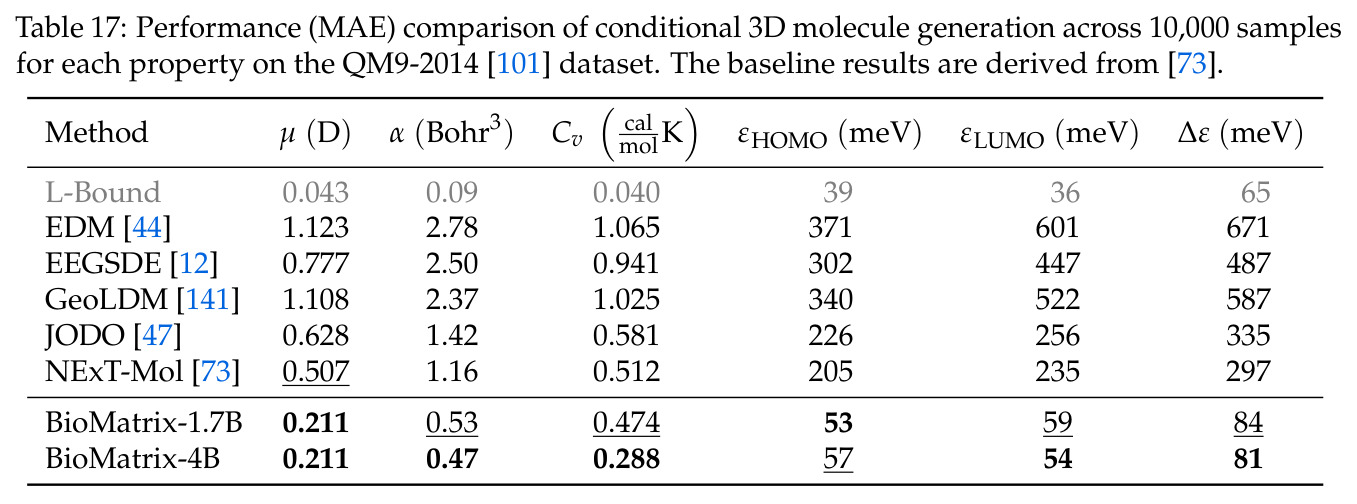
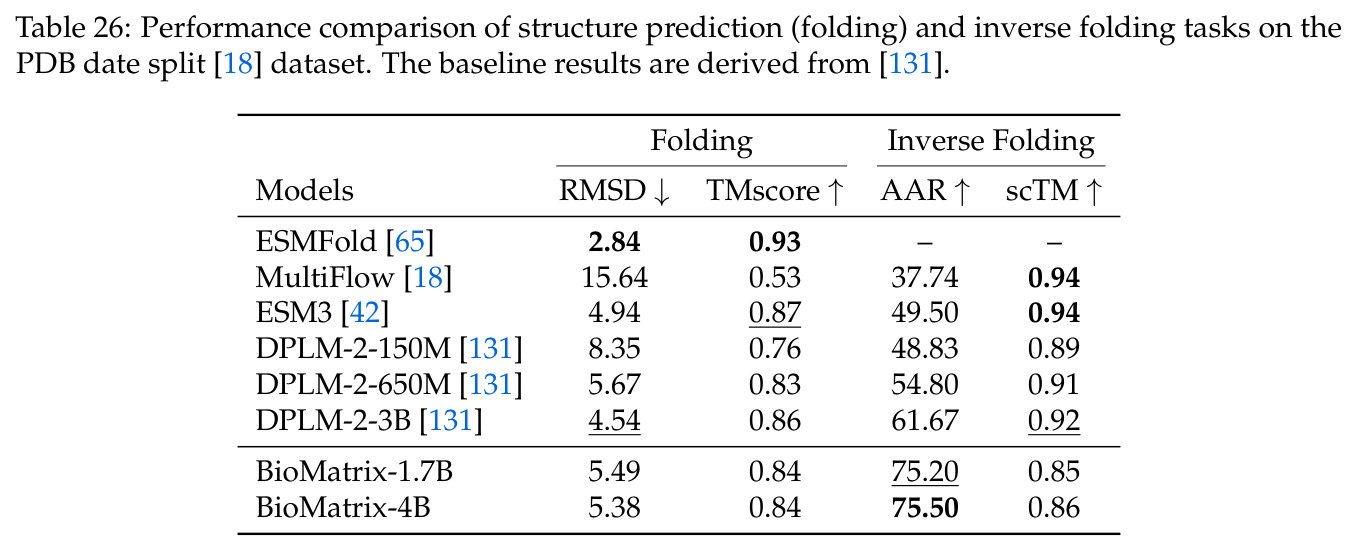
查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| 无条件分子生成 (MOSES) | Validity | 0.998 | OMG-GPT 0.990 | +0.008 |
| 文本基分子生成 | Exact Match | 65.07% | SciReasoner-8B 48.00% | +17.07% |
| 属性条件 3D 分子生成 (HOMO) | MAE (meV) | 53 | NExT-Mol 205 | -74.1% |
| EC Number Prediction (Price) | F1 | 34.34% | SciReasoner-8B 22.00% | +12.34% |
| 蛋白质反向折叠 | Amino Acid Recovery | 75.50% | DPLM-2-3B 61.67% | +13.83% |
| 序列-结构联合生成 (struct→seq) | self-consistency TM | 0.979 | DPLM-2-650M 0.921 | +0.058 |
| 分子-蛋白质亲和力 (PDBBindv2020) | RMSE | 1.260 | TankBind 1.346 | -6.4% |
| 分子-蛋白质亲和力 (BindingDB) | RMSE | 1.030 | Literature SOTA 1.340 | -23.1% |
| 分子知识问答 (MoleculeQA) | Total Accuracy | 73.78% | MolCA-1.3B 64.79% | +8.99% |
| 属性条件分子优化 (logP) | Success Rate | 0.9838 | Llama3.1-8B 0.8822 | +0.1016 |
局限与改进
作者承认的主要限制包括:分子和蛋白质的 3D 结构 tokenizer 使用不相交的几何参考系,无法原生表示小分子和蛋白质口袋之间的相对位姿,因此无法表示或生成生物分子复合物,限制了结构对接和口袋条件配体设计等任务的应用;持续预训练语料库与下游 SFT/评估数据之间可能存在实体重叠,因为分子和蛋白质在数据库和基准测试中被重复使用;领域专业化导致通用能力部分退化,尽管加入了通用科学文本,但 3044 亿 token 中生物分子 token 的高浓度不可避免地使模型偏离 Qwen3-Base 的通用语言和推理能力;采用任务分组微调而非单一统一模型,因为生物 SFT 数据的模式单一性导致大数据子任务主导小数据子任务;当前仅覆盖小分子和蛋白质,不包括核酸、碳水化合物和脂质等其他重要生物实体。作者还指出 3D 几何任务的瓶颈在于 tokenizer 而非建模能力:QM9-2014 的键长 MMD 相对宽松,折叠 RMSD 落后于 ESMFold,但轻量级的 MMFF 优化可以缩小大部分差距(FCD:1.04→0.23,atom stability:0.897→0.985)。
独立分析的弱点
BioMatrix 的主要弱点集中在三个方面。第一,3D 几何重建的精度受限于 tokenizer 的量化误差和自回归重建的误差累积。在 QM9-2014 上,原始构象的键长 MMD 为 1.05,明显宽松于 NExT-Mol,虽然 MMFF 优化可以改善,但这增加了后处理步骤。改进方向是采用分层 codebook 或非自回归解码方案,如并行重建所有原子坐标。第二,无法表示生物分子复合物的相对位姿,因为分子和蛋白质的 tokenizer 使用不相交的参考系。改进方向是设计一个跨实体类型的统一 3D tokenizer,使用共享的全局参考系或显式的相对位姿编码。第三,任务分组微调而非统一模型导致实际使用时需要在多个变体间选择。改进方向是探索更 principled 的数据平衡策略、课程设计或多任务生物语料库的专门训练动力学。
未来方向
作者提出的未来方向包括:开发统一的 3D tokenizer 以支持跨实体类型的复合物生成,这将使结构对接和口袋条件配体设计等任务成为可能;将覆盖范围扩展到核酸、碳水化合物和脂质等其他生物实体,与现有的框架概念兼容;解决小数据子任务在统一 SFT 中被大数据子任务主导的问题,可能通过更智能的数据平衡、课程学习或任务特定正则化;改进结构 tokenizer 以支持更高精度的几何重建,包括侧链建模和更细粒度的码本。基于本文成果,延伸方向包括:探索统一 token 空间在生物分子动力学模拟中的应用,将时间维度作为额外的模态;研究如何将 BioMatrix 与实验设计工作流集成,实现从文本描述到实验验证的端到端流程;开发专门的推理优化技术,针对生物分子任务的特定模式进行剪枝或蒸馏,降低部署成本。
复现评估
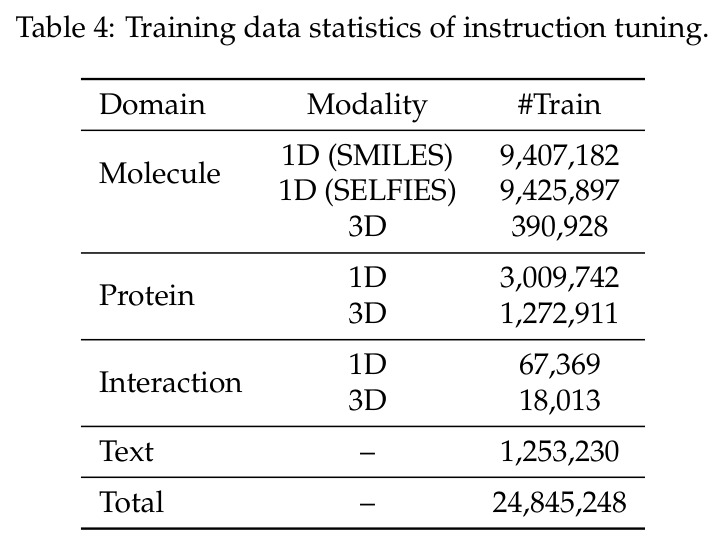
BioMatrix 的代码和模型已开源(GitHub: QizhiPei/biomatrix,HuggingFace: QizhiPei/biomatrix),但完整复现成本较高。持续预训练使用了 3044 亿 token 的语料库,在 64 张 NVIDIA H100 GPU 上训练约 36400 步,全局 batch size 为 1024,最大序列长度为 8192 token,使用 AdamW 优化器,峰值学习率为每批 2.0 乘 10 的负 4 次方,cosine 调度,2000 步 warmup。这对于大多数研究实验室来说是巨大的算力投入。预训练语料库来源包括公开数据库(PubChem、UniProt、AlphaFoldDB 等)和文本语料(FineWeb-Edu、PubMed 等),但这些数据需要大量的清洗和处理工作。指令微调的算力需求较低,但需要准备 2480 万训练样本。简化版本的复现(如仅分子任务或仅蛋白质任务)可以在较小规模的数据集和较少的 GPU 上进行,但性能会有所下降。论文提供了详细的训练配置和数据来源说明,但没有提供数据预处理脚本,这是复现的一个潜在障碍。
论文图表