CellMaster:单细胞分析中的协作式细胞类型注释框架 CellMaster: Collaborative Cell Type Annotation in Single-Cell Analysis
基于LLM的多智能体系统,通过迭代推理实现零样本单细胞类型注释
前置知识
单细胞RNA测序(scRNA-seq)
单细胞RNA测序是一种高通量测序技术,能够在单个细胞水平上测量基因表达谱。与传统批量测序不同,scRNA-seq可以揭示组织中不同细胞类型的异质性,识别稀有细胞群和瞬态细胞状态。该技术通过将组织解离成单细胞、逆转录mRNA、构建cDNA文库并进行高通量测序来实现,最终产生包含每个细胞数千个基因表达水平的表达矩阵。
理解scRNA-seq是本文的核心应用场景,CellMaster正是为解决scRNA-seq数据的细胞类型注释问题而设计的
细胞类型注释
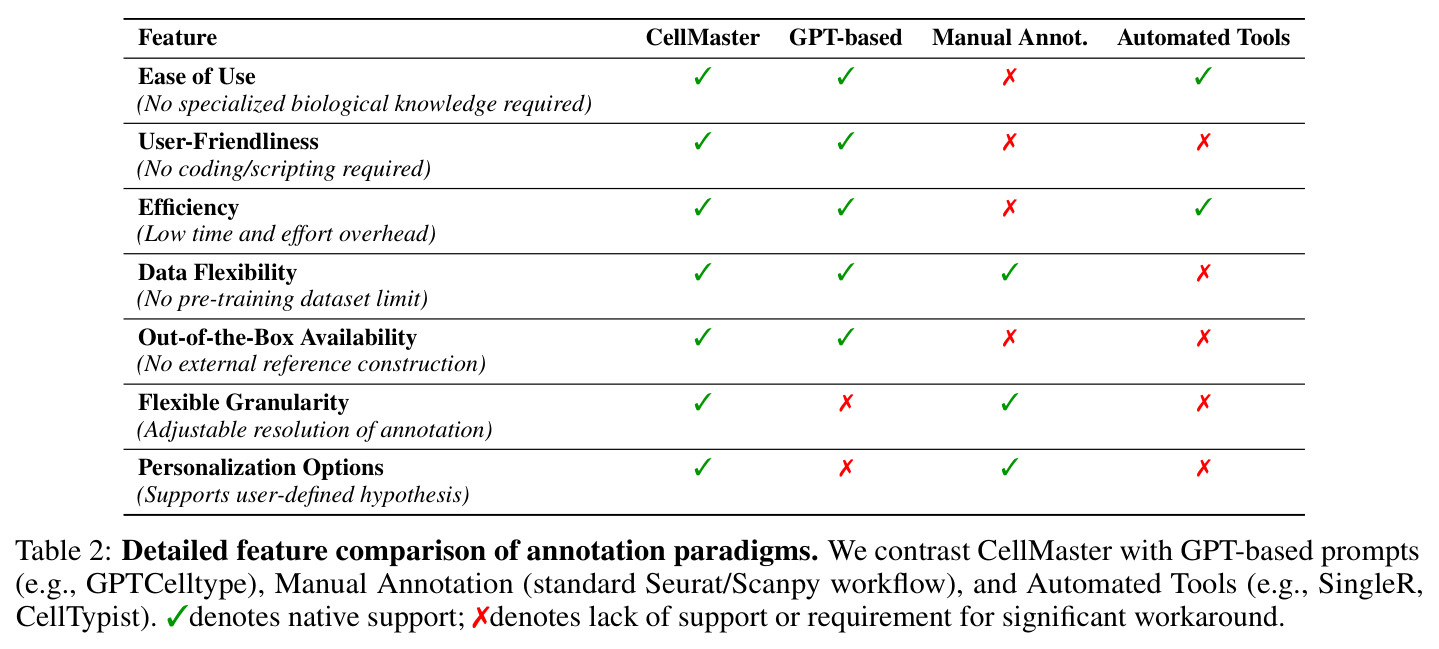
细胞类型注释是将scRNA-seq数据中的细胞聚类分配到已知生物学细胞类型标签的过程。传统方法依赖人工专家通过分析差异表达基因(DEGs)、标记基因和生物学背景来手动注释,这个过程耗时且难以扩展。自动化方法包括标记基因匹配(如CellMarker 2.0)、参考标签转移(如Seurat)和监督分类(如CellTypist、scTab)。
这是CellMaster要解决的核心任务,理解现有注释方法的局限性是理解本文动机的关键
大语言模型(LLM)
大语言模型是基于Transformer架构的深度学习模型,通过在海量文本数据上预训练获得强大的语言理解和推理能力。代表性模型包括GPT-4o、Gemini等。在生物医学领域,LLM能够编码大量生物学知识,包括基因功能、细胞类型特征和分子通路信息,使其能够在没有专门训练的情况下执行特定领域的推理任务。
CellMaster的核心创新在于利用LLM的推理能力来模拟专家的注释决策过程,实现零样本注释
多智能体系统
多智能体系统是一种由多个协作智能体组成的架构,每个智能体负责特定任务,通过协调和通信完成复杂目标。在CellMaster中,系统包含四个专门阶段的智能体:假设生成、标记选择、表达分析和结果评估。这种模块化设计使得系统能够进行迭代推理,在每一步产生可解释的中间结果。
多智能体架构是CellMaster区别于单次LLM调用方法(如GPTCelltype)的关键技术特征
人机协作(Human-in-the-loop)
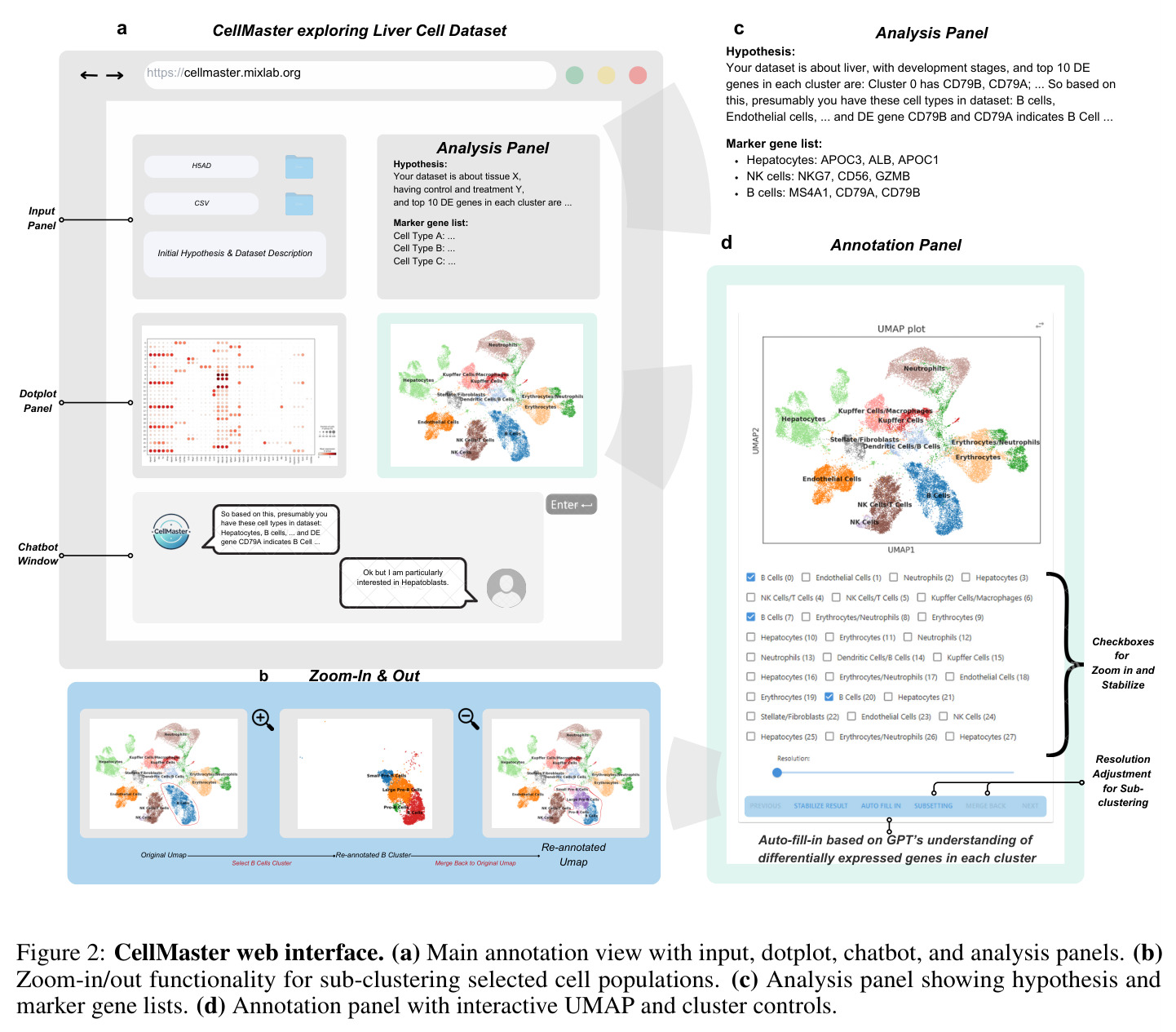
人机协作是一种设计范式,允许人类专家在AI系统的决策过程中进行干预和指导。在CellMaster中,专家可以通过Web界面审查LLM的推理过程、提供反馈、调整注释粒度,并标记不确定的聚类。系统会记录所有人工干预的来源,确保可追溯性。这种方式结合了AI的计算能力和人类的生物学直觉。
人机协作模式使CellMaster的性能提升了18.6%,是本文的重要贡献之一
差异表达分析
差异表达分析是识别在不同细胞群体之间表达水平存在显著差异的基因的统计方法。在单细胞分析中,通常使用Wilcoxon秩和检验来比较每个聚类与其他聚类的基因表达差异,识别每个聚类的标记基因。这些标记基因是细胞类型注释的基础,因为不同细胞类型通常具有特征性的基因表达模式。
CellMaster的假设生成阶段依赖差异表达分析结果来驱动迭代推理过程
研究动机
单细胞RNA测序(scRNA-seq)技术的发展使得大规模组织图谱分析成为可能,但细胞类型注释仍然是下游分析和跨研究整合的核心瓶颈。传统的人工注释方法虽然能产生高质量标签,但耗时且难以扩展到包含数百万细胞的现代图谱数据集。现有的自动化方法存在显著局限性:标记基因匹配方法(如CellMarker 2.0)依赖于预定义的标记数据库,无法处理缺乏共识标记的稀有或新细胞状态;参考标签转移方法(如Seurat)对批次效应和领域偏移敏感;监督分类方法(如CellTypist和scTab)在处理参考数据覆盖不足的组织时表现不佳,特别是CellTypist在注释少于100个细胞的聚类时性能显著下降。更重要的是,这些方法的决策过程缺乏生物学可解释性,难以在探索性研究中建立信任。近期LLM方法如GPTCelltype虽然展示了潜力,但仅执行单次推理,缺乏迭代优化和人机交互能力。
本文的目标是本文的目标是开发一个能够模拟专家推理过程的AI系统CellMaster,实现零样本、可解释的细胞类型注释。具体而言,CellMaster旨在:(1)通过LLM编码的生物学知识进行即时注释,无需预训练或固定标记数据库;(2)提供可解释的推理过程,让专家能够理解和验证注释决策;(3)支持迭代精炼,通过假设生成、标记选择、表达分析和结果评估的循环不断改进注释质量;(4)实现人机协作,允许专家在注释过程中提供指导和反馈;(5)在稀有细胞群和新细胞状态上表现出色,这些场景下现有方法往往失败。系统最终目标是在9个数据集、8种组织类型上超越现有最佳基线方法。
与已有工作不同的是,CellMaster的独特切入角度在于将LLM的推理能力与专家工作流程深度整合,而非简单地将LLM用作黑盒分类器。与现有方法的本质区别体现在三个方面:首先,CellMaster采用迭代推理框架,而非单次预测,通过多轮假设-验证循环逐步精炼注释,这模拟了人类专家的实际工作方式;其次,系统设计强调可解释性,每一步推理都产生自然语言推理理由、点图可视化和置信度分数,使注释过程完全透明;第三,CellMaster构建了完整的人机协作界面,允许专家在推理过程中进行干预,包括审查中间结果、调整标记基因、改变注释粒度和提供文本反馈。这种开放式设计将LLM从黑盒自动化工具转变为响应式研究伙伴,使计算推理与专家生物学直觉对齐,这在现有方法中尚未实现。
核心方法
CellMaster的核心思路是将专家的细胞类型注释工作流程分解为四个专门的推理阶段,并通过LLM驱动的多智能体架构实现迭代精炼。整体方法遵循一个直觉:专家注释时会先提出假设(这个聚类可能是什么细胞类型),然后选择验证这些假设的标记基因,接着检查这些标记的表达模式,最后评估结果并决定是否需要进一步分析。CellMaster将这个过程形式化为四个阶段的循环:假设生成(Hypothesis Generation)→标记选择(Marker Selection)→表达分析(Expression Analysis)→结果评估(Result Evaluation)。技术路线基于Scanpy预处理流程,首先对scRNA-seq数据进行标准化和对数变换,然后使用Wilcoxon秩和检验识别每个聚类的差异表达基因。系统使用GPT-4o作为基础模型,通过精心设计的提示词(prompts)驱动每个阶段的推理。在自动模式下,系统默认运行3次迭代,未解决的聚类使用"自动填充"功能处理。在人机协作模式下,专家可以通过Web界面在每次迭代中提供最多3次输入,审查推理理由并指导后续分析方向。
CellMaster的核心创新在于将LLM从简单的标签预测器转变为能够进行假设驱动推理的自主代理。与GPTCelltype等方法仅根据前10个标记基因进行一次性预测不同,CellMaster实现了完整的专家推理循环。关键区别体现在:(1)迭代精炼机制——系统在每次迭代中不仅注释新聚类,还会重新审视之前的结果,通过新增的标记基因和表达证据不断精炼;(2)标记记忆系统——系统维护成功和失败标记基因的动态列表,避免重复使用无效标记,自适应地调整标记选择策略;(3)置信度稳定化——通过多轮评估产生置信度分数,识别需要进一步分析的不确定聚类;(4)上下文感知推理——系统利用数据集的生物学背景(如发育阶段、组织类型)来解释标记基因表达模式,而非仅依赖通用标记知识。这些机制使CellMaster能够处理现有方法难以应对的挑战场景,如标记基因冲突(如Cd3d vs Nkg7的区分)、发育背景利用(如Afp作为肝母细胞标记 vs Alb作为肝细胞标记)和调控子支持的决策。
方法步骤详情
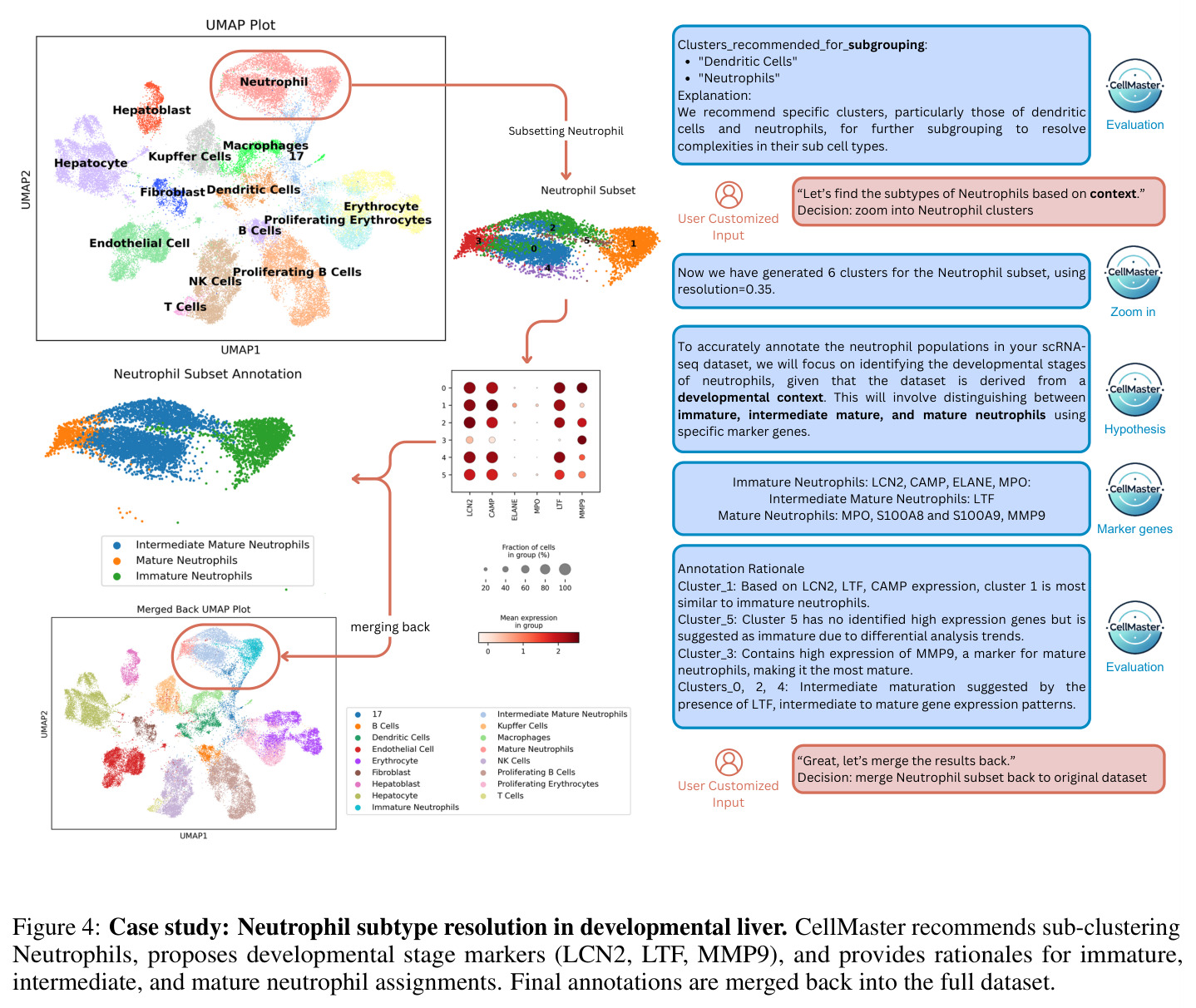
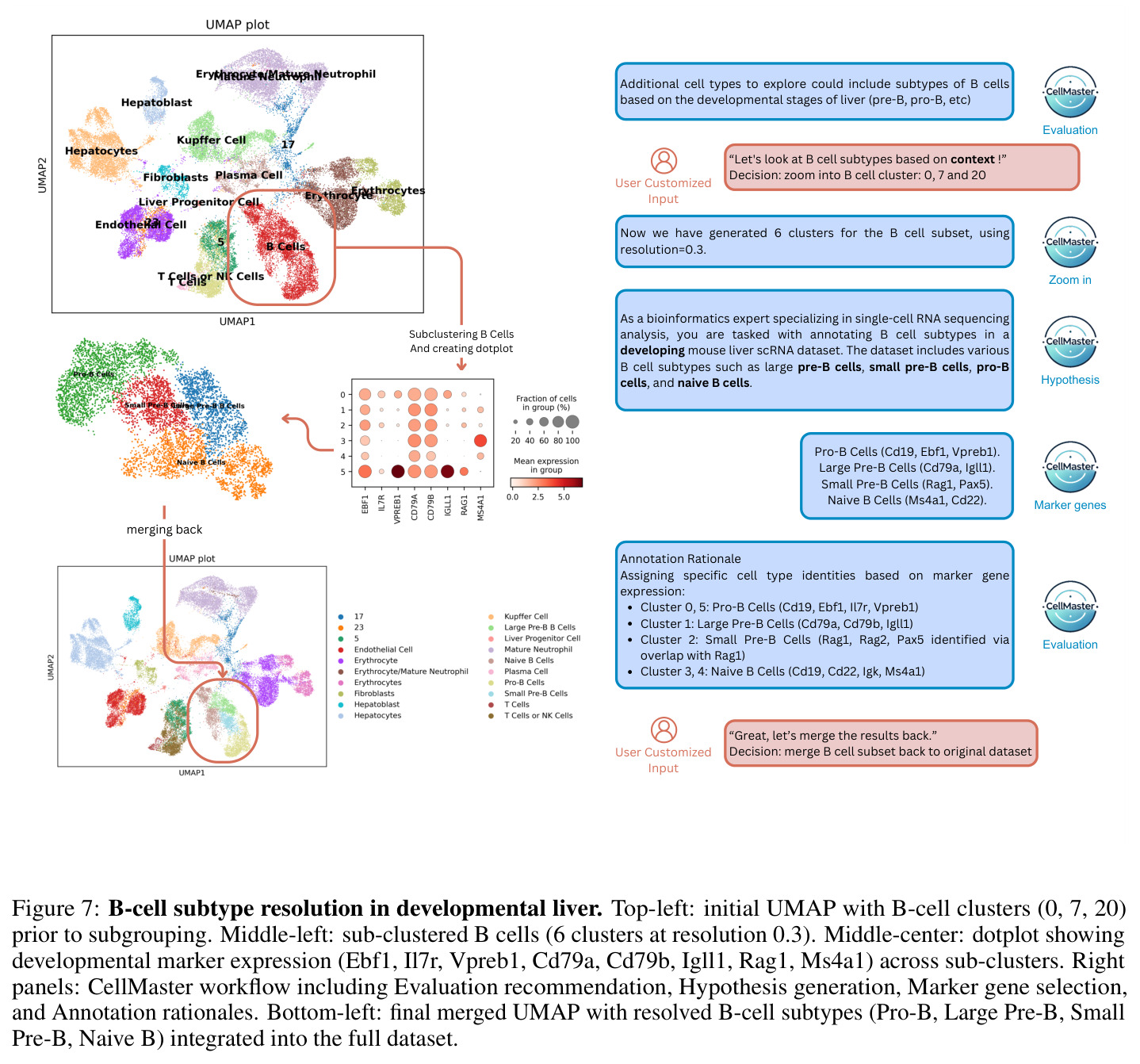
CellMaster的迭代注释流程包含四个专门阶段,每个阶段都有明确的输入输出。第一阶段是假设生成(Hypothesis Generation):输入为差异表达分析结果(每个聚类的前N个高平均log2倍数变化基因)和之前的注释结果,输出为关于细胞类型分布的生物学假设。系统分析DE基因模式,结合数据集背景,提出每个聚类可能的细胞类型候选。第二阶段是标记选择(Marker Selection):输入为当前假设和之前迭代的标记记忆(成功/失败标记列表),输出为用于区分特定细胞类型的标记基因列表。系统动态维护标记记忆,避免重复使用无效标记,战略性地选择能够解决不确定聚类的标记。第三阶段是表达分析(Expression Analysis):输入为选定的标记基因和原始表达数据,输出为可视化点图(dotplots),展示基因表达在聚类间的分布。点图通过点大小表示表达细胞比例,颜色强度表示表达水平,验证标记基因的相关性和区分能力。第四阶段是结果评估(Result Evaluation):输入为表达分析结果,输出为多层面评估,包括基因特异性、聚类标记签名、相似聚类对识别、置信度分数和后续分析建议。评估结果动态保存,注释后的h5ad对象和UMAP可视化自动生成。系统还集成了多种自适应启发式规则,包括标记记忆、置信度稳定化、聚类关系分析和污染检测规则。
技术新颖性
CellMaster的技术新颖性体现在多个维度。首先,在架构设计上,CellMaster是首个将多智能体LLM架构应用于单细胞注释的系统,通过四个专门化的推理阶段实现模块化的专家工作流模拟,这与CASSIA等多智能体方法的"流水线"式设计不同,CellMaster强调迭代反馈和上下文传递。其次,在推理机制上,系统实现了标记记忆和置信度稳定化,这是现有LLM注释方法所不具备的。标记记忆使系统能够从失败中学习,避免重复错误;置信度稳定化通过多轮评估产生可靠的不确定性估计。第三,在人机交互设计上,CellMaster构建了完整的协作界面,支持来源追踪的编辑历史,所有人工干预都有记录,确保可审计性。这与Biomni等通用生物AI工具的"一次性"交互模式形成对比。第四,在评估框架上,本文开发了基于Gene Ontology的相似性评分体系,采用分层注释结构和本体映射,实现了生物学意义的跨方法比较。这种评估方法考虑了细胞类型的层级关系(如T细胞→CD4 T细胞→调节性T细胞),比简单的标签匹配更能反映注释质量。
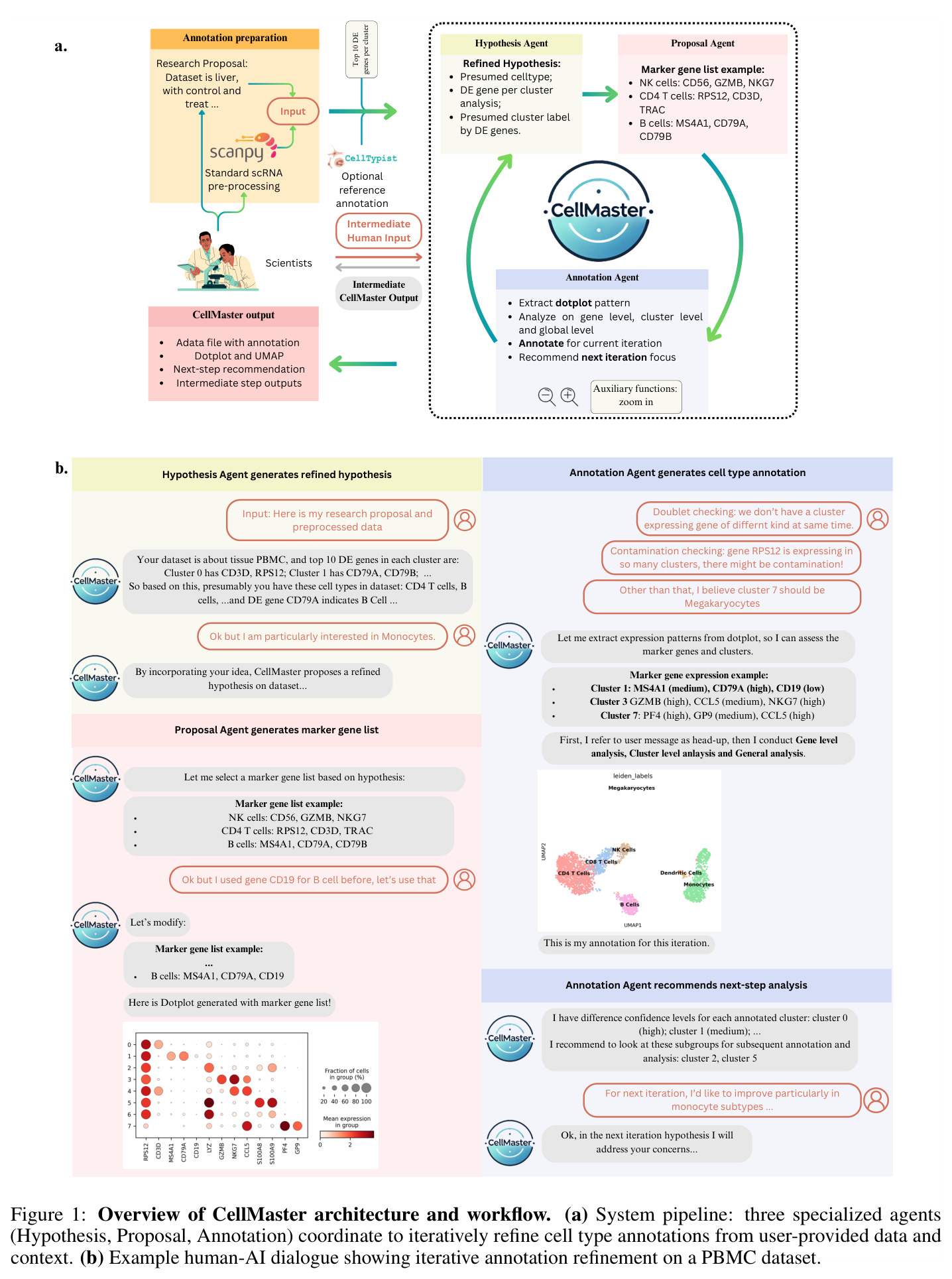
实验结果
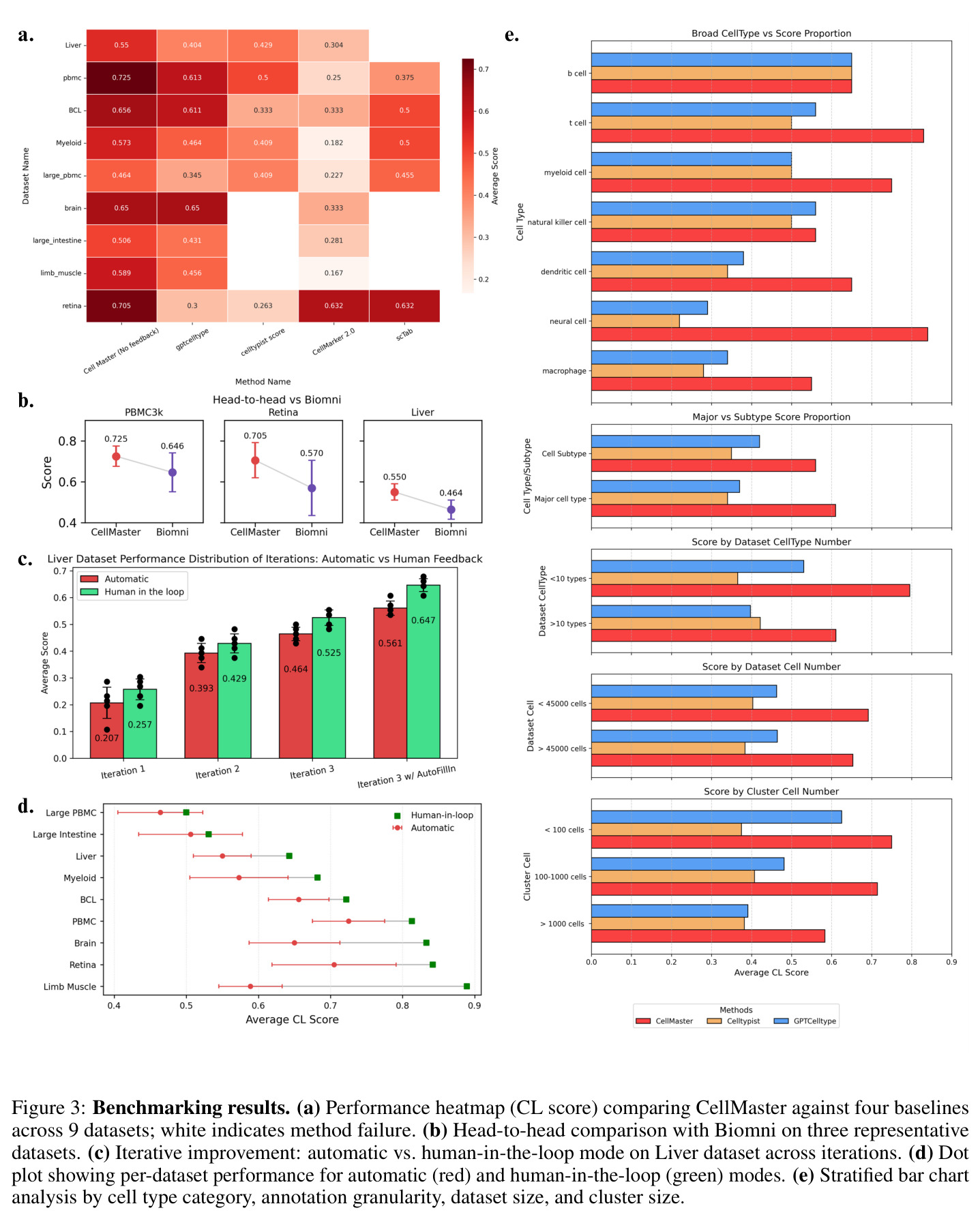
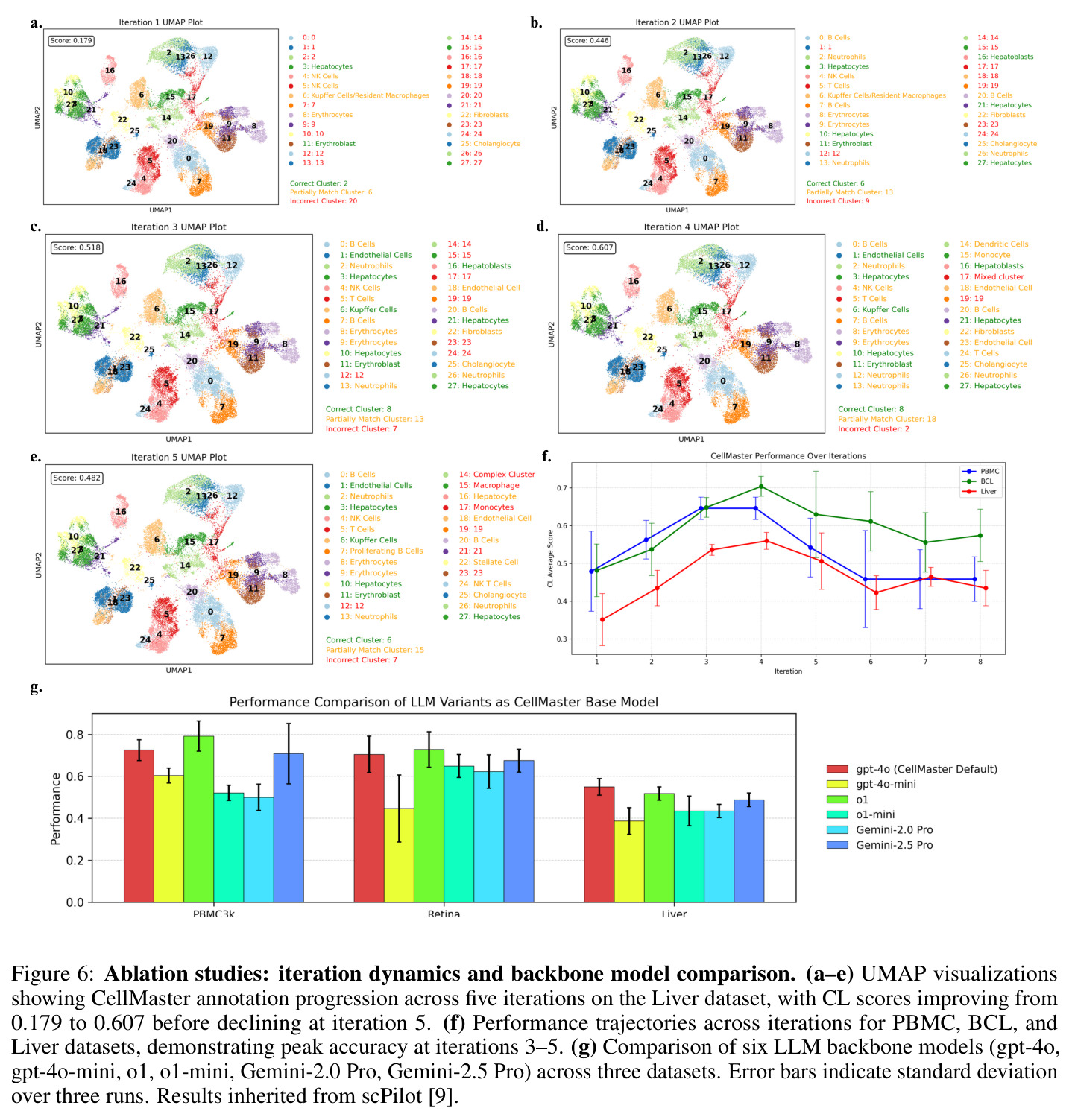
CellMaster在9个数据集、8种组织类型上的全面评估展示了其卓越性能。在自动模式下,CellMaster实现了0.602±0.058的平均性能分数,比每个数据集的最佳基线方法平均高出0.071(约13%的相对提升)。特别值得注意的是在Retina数据集上(0.705 vs 0.300-0.632)和Liver数据集上(0.55 vs 0.304-0.429)取得的显著改进。在人机协作模式下,性能优势进一步扩大至0.115±0.085的平均提升,超越所有基准方法。在亚细胞类型注释方面,CellMaster比基线方法高出22.1%,展示了其在精细粒度注释上的优势。与Biomni(基于Gemini-2.5 Pro)的对比显示,CellMaster在PBMC3k、Retina和Liver数据集上分别获得0.725、0.705和0.550的分数,而Biomni分别为0.646、0.570和0.464,且CellMaster的变异性更低(标准差≤0.086 vs Biomni的最高0.135)。消融实验表明,GPT-4o在所有数据集上提供了平衡的基线性能,而o1模型在PBMC3k和Retina上有所提升但在Liver上下降,表明可能存在过度推理问题。迭代性能分析显示,Liver数据集的性能从第1次迭代的0.179单调提升至第4次迭代的0.607,但在第5次迭代下降至0.482,揭示了自动模式下的性能平台效应。

查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| 自动模式细胞类型注释(9个数据集平均) | GO感知相似性分数(CL score) | 0.602±0.058 | 各数据集最佳基线平均0.531 | 提升0.071(约13%相对提升) |
| 人机协作模式注释 | 相比自动模式的性能提升 | 0.717(自动模式0.602+提升0.115) | 自动模式CellMaster 0.602 | 提升0.115±0.085 |
| 亚细胞类型注释 | 与基线方法的性能差距 | 显著优于GPTCelltype和CellTypist | GPTCelltype和CellTypist | 提升22.1% |
| Retina数据集注释 | CL score | 0.705 | 0.300-0.632(基线范围) | 提升0.073-0.405 |
| Liver数据集注释 | CL score | 0.550 | 0.304-0.429(基线范围) | 提升0.121-0.246 |
| vs Biomni对比(PBMC3k) | CL score | 0.725 | Biomni 0.646 | 提升0.079 |
| vs Biomni对比(Retina) | CL score | 0.705 | Biomni 0.570 | 提升0.135 |
| vs Biomni对比(Liver) | CL score | 0.550 | Biomni 0.464 | 提升0.086 |
局限与改进
论文作者坦诚地承认了CellMaster的几个主要局限性。首先,LLM的概率性质引入了随机性,导致运行间变异性略高于简单基线方法(平均标准差0.058 vs GPTCelltype的0.053),尽管有稳定化机制但仍然存在。其次,对商业模型API的依赖引发了成本和隐私问题,特别是在受监管的研究环境中,这限制了系统的可访问性和数据安全性。第三,基于Cell Ontology映射的评估方法可能存在偏差,对真正新颖的、不存在于现有层级中的细胞状态不利,这意味着评估框架可能低估了CellMaster在发现新细胞类型方面的能力。第四,当前框架仅限于单模态转录组学数据,未整合多组学信息。从独立观察来看,系统在自动模式下存在性能平台效应,3-5次迭代后性能会稳定甚至下降,这表明LLM在没有人类指导时容易过度自信或产生不一致的推理。此外,系统对基础模型的选择敏感,不同LLM在不同数据集上表现差异较大,缺乏统一的最优模型选择策略。
独立分析的弱点
尽管CellMaster取得了显著成果,但存在几个值得深入分析的弱点。首先,迭代性能下降问题:在Liver数据集上,第5次迭代的性能从0.607下降至0.482,系统会重新考虑已解决的聚类并替换关键标记基因(如丢失Afp导致肝母细胞被误标为肝细胞)。改进方向包括实现保守的早期停止策略,或在迭代过程中锁定高置信度注释。其次,标记记忆的局限性:虽然系统维护成功/失败标记列表,但在复杂场景下(如标记基因冲突),记忆机制可能不足以避免重复错误。可以考虑引入更复杂的标记关系图谱或基于知识图谱的标记验证。第三,人机协作的效率瓶颈:每次迭代仅允许3次人工输入,且界面设计可能对非计算背景的生物学家不够友好。改进方向包括开发更直观的可视化交互和智能建议系统。第四,评估框架的局限性:基于本体映射的评分可能无法充分评估对新细胞状态的发现能力,需要开发专门针对新颖性发现的评估指标。第五,计算效率:多轮LLM推理的累积成本较高,特别是在大规模数据集上,需要优化提示词设计或引入模型蒸馏策略。
未来方向
论文作者提出了几个明确的未来研究方向。首先是减少随机性:通过约束解码(constrained decoding)技术来提高LLM输出的一致性,这可以通过限制词汇表或强制输出结构来实现。其次是扩展本体覆盖:当前基于Cell Ontology的评估可能限制了对新兴细胞类型的识别,需要建立更全面的细胞类型本体体系。第三是多模态数据整合:将框架扩展到支持scATAC-seq和空间转录组学数据,这将极大地扩展系统的应用范围。基于当前成果,还可以延伸以下方向:(1)开发专门针对稀有细胞发现的评估基准,超越简单的注释准确性;(2)构建跨数据集的知识迁移机制,使系统能够从一个组织类型的学习经验中受益于其他组织;(3)实现自适应的迭代策略,根据聚类复杂度动态调整迭代次数和标记选择策略;(4)开发轻量级模型版本,降低对商业API的依赖,使系统能够在本地部署;(5)整合主动学习策略,智能地选择最需要人类干预的聚类,提高人机协作效率。
复现评估
CellMaster在可复现性方面表现良好。代码完全开源,以MIT许可证发布在GitHub上(https://github.com/AnonymousGym/CellMaster),包含完整的源代码、数据集预处理流程、评估指标实现和基准测试驱动程序。所有使用的scRNA-seq数据集都是公开可获取的集合,论文详细列出了数据来源。系统基于广泛使用的Scanpy预处理流程构建,降低了技术门槛。然而,复现存在一些挑战:首先,系统依赖商业LLM API(主要是GPT-4o),需要有效的API密钥和相应的使用成本,这可能限制了在资源有限的研究组中的可复现性。其次,人机协作模式的评估涉及主观的人工反馈,完全复现需要相同的专家知识和判断。第三,虽然论文提供了详细的提示词描述(Supplement G),但LLM的输出可能因模型版本更新而有所变化。总体而言,对于自动模式的基准测试,复现难度为中等;对于人机协作模式的完整评估,复现难度较高。算力需求方面,主要成本在于LLM API调用,无需专门的GPU硬件。
论文图表