scPilot:面向自动化单细胞分析与发现的大语言模型推理框架 scPilot: Large Language Model Reasoning Toward Automated Single-Cell Analysis and Discovery
首个系统性LLM原生推理框架,自动化单细胞组学分析
前置知识
单细胞RNA测序(scRNA-seq)
单细胞RNA测序是一种在单个细胞层面测量基因表达水平的技术。与传统的批量RNA测序不同,scRNA-seq能够捕获每个细胞独特的转录组特征,生成一个基因×细胞的大规模表达矩阵 X ∈ R^{G×N},其中 G 为基因数(通常数万),N 为细胞数(通常数万到数百万)。该数据是后续细胞类型注释、发育轨迹重建等分析的基础。
SCPILOT处理的核心数据就是scRNA-seq表达矩阵,理解其结构和挑战(高维、稀疏、噪声大)是理解本文问题设定的前提。
细胞类型注释(Cell Type Annotation)
细胞类型注释是scRNA-seq分析中最基础的任务,目标是根据基因表达模式为每个细胞分配生物学上有意义的标签(如T细胞、B细胞、NK细胞等)。传统方法依赖人工查阅文献标记基因(marker genes),自动化工具如CellTypist基于预训练分类器,但准确率有限,尤其是在组织特异性细胞类型上。
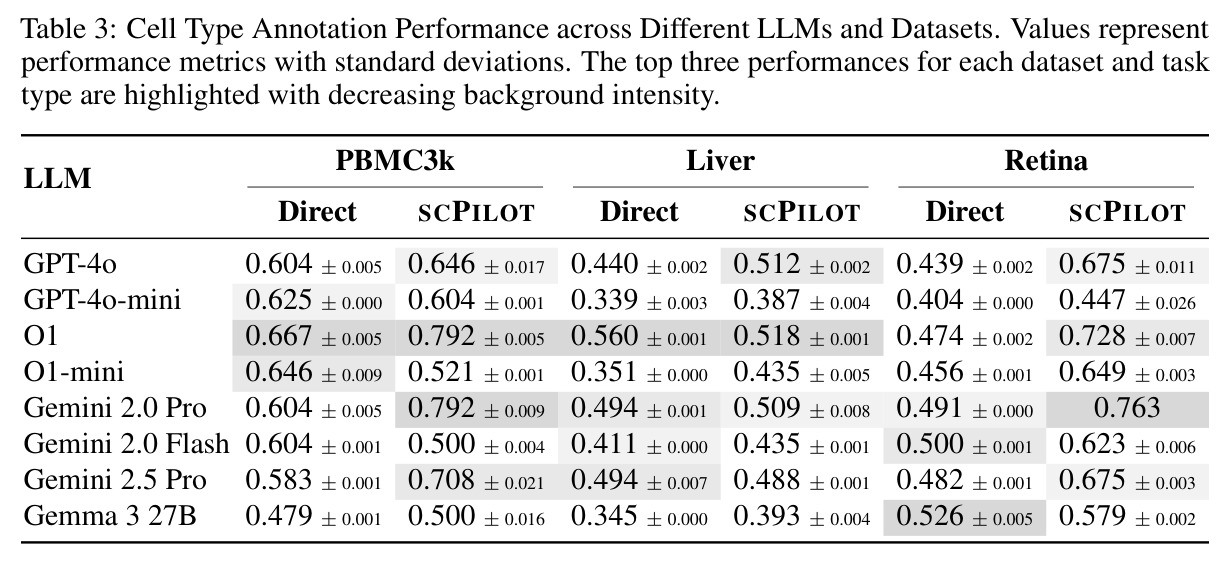
这是SCPILOT框架评估的三大核心任务之一,论文中用三个数据集(PBMC3k、Liver、Retina)系统评估了该任务的性能。
发育轨迹推断(Trajectory Inference)
发育轨迹推断旨在从scRNA-seq数据中重建细胞的发育或分化路径,通常以谱系树(lineage tree)的形式表示。Monocle 3等工具通过计算伪时间(pseudotime)和细胞间连接来推断轨迹结构。该任务的核心挑战在于准确识别分支点和分化方向。
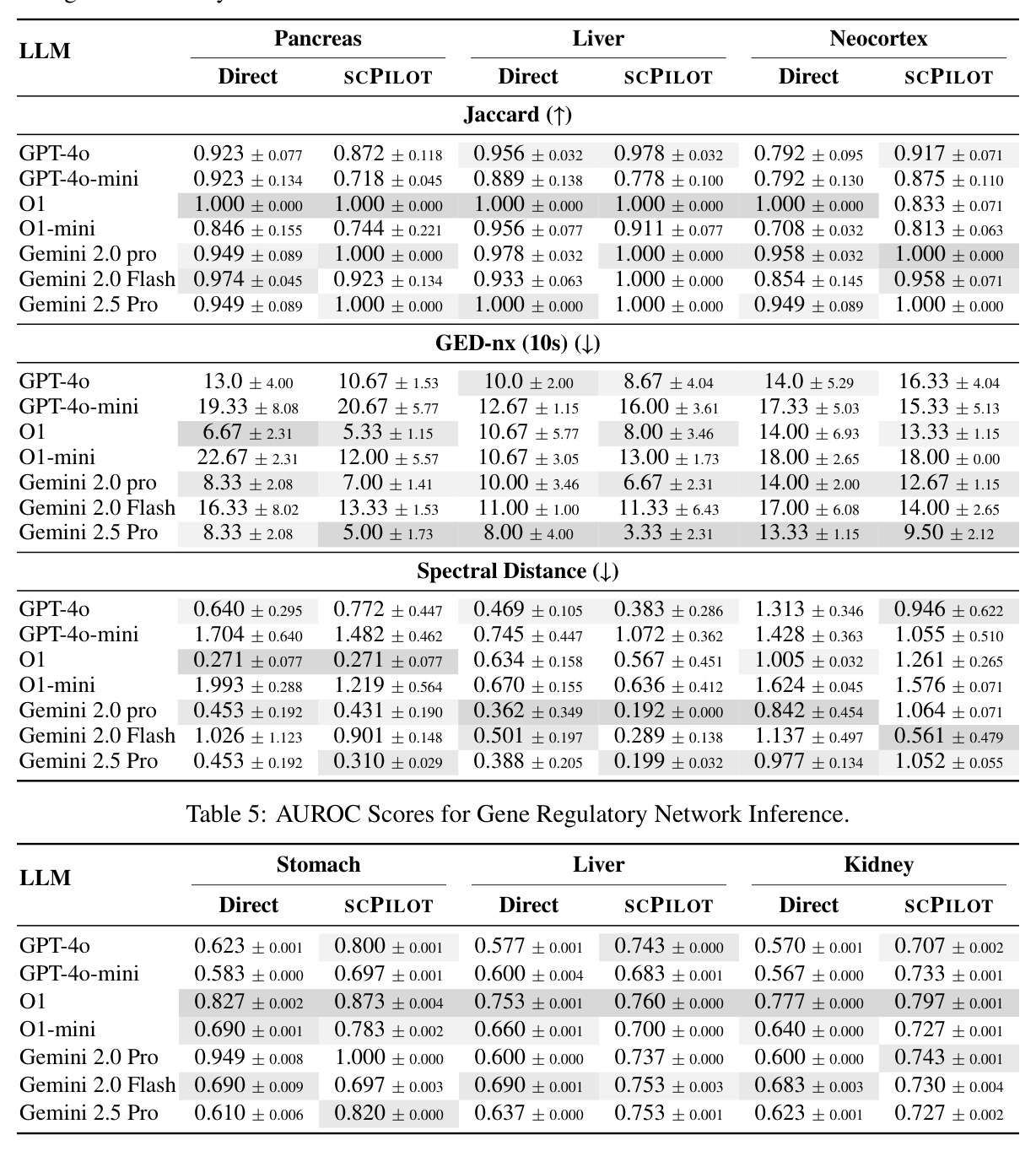
SCPILOT将轨迹推断作为第二大评估任务,使用Pancreas、Liver和Neocortex三个数据集,并引入图编辑距离(GED)等结构化评估指标。
基因调控网络(Gene Regulatory Network, GRN)
基因调控网络描述转录因子(Transcription Factor, TF)对靶基因的调控关系,通常表示为有向图。SCENIC等工具通过基序分析和共表达推断TF-基因对,然后通过实验验证(如TRRUST数据库)进行确认。预测TF-基因调控关系对于理解细胞命运决定和疾病机制至关重要。
GRN预测是SCPILOT的第三大任务,论文使用GRNdb数据集和TRRUST验证集进行评估,SCPILOT的AUROC从直接提示的0.827提升到0.873。
Omics-Native Reasoning(ONR,组学原生推理)
ONR是本文提出的核心概念,指大语言模型直接在组学数据空间中进行推理的范式。与传统工具调用不同,ONR要求LLM:(i) 接收单细胞数据的文本摘要,(ii) 明确提出生物学假设,(iii) 直接调用生物信息学工具获取证据,(iv) 评估数值证据,(v) 迭代修正推理直至得出结论。整个推理过程记录为可审计的推理轨迹。
这是SCPILOT的核心创新,将LLM从简单的代码生成器转变为真正的生物学推理伙伴,论文的所有实验都基于这一范式。
图编辑距离(Graph Edit Distance, GED)
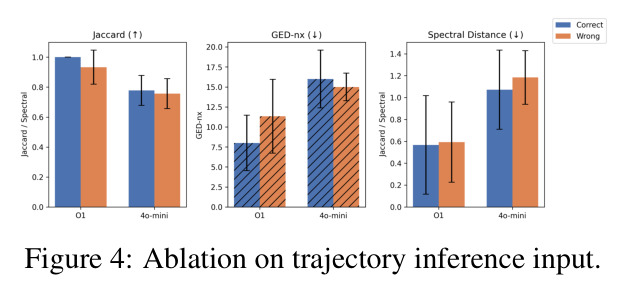
图编辑距离是衡量两个图结构差异的度量,定义为将一个图转换为另一个图所需的最小编辑操作(添加/删除节点或边)数量。在SCBENCH中,GED-nx用于评估推断的轨迹树与专家标注的ground truth谱系树之间的结构差异,数值越低表示推断越准确。
GED是轨迹推断任务的核心评估指标之一,论文报告Gemini-2.5-Pro将GED降低了30%,这是SCPILOT最重要的性能提升之一。
研究动机
单细胞组学数据的爆炸式增长使得传统的分析流程面临严峻挑战。现代scRNA-seq实验可产生百万级细胞的表达矩阵,但分析流程仍然严重依赖研究者的隐式经验和主观判断。具体而言,细胞类型注释需要专家手动查阅文献中的标记基因,Monocle和SCENIC等工具暴露大量超参数和不透明的默认设置,Web平台(如ASAP)和一键框架(如SPEEDI)虽然简化了执行流程,但仍嵌入了在新组织或新扰动条件下可能失效的刚性启发式规则。现有的LLM工具代理(如CellAgent使用GPT-4自动选择工具和超参数)虽然令人印象深刻,但主要封装了默认启发式方法,对工具调用背后的生物学洞察有限。基础模型(如scGPT等)将单细胞计数嵌入到不透明的高维向量空间中,虽然在嵌入质量上表现良好,但缺乏关键的生物学可解释性。简而言之,当前的方法要么是黑箱式的向量表示,要么是包装固定结果的聊天接口,都无法满足单细胞分析对透明推理和生物学发现的需求。
本文的目标是本文的具体目标是创建一个系统性的框架,使大语言模型能够像人类生物学家一样直接在原始单细胞数据上进行推理。具体来说,SCPILOT旨在实现以下目标:(1) 将细胞类型注释、发育轨迹重建和转录因子靶标预测这三大核心单细胞分析任务转化为LLM可以逐步解决、论证和修正的推理问题;(2) 生成透明的推理过程,能够解释标记基因歧义和调控逻辑;(3) 通过迭代推理显著提升分析准确率,在细胞类型注释上提升11%平均准确率,轨迹推断中降低26%的图编辑距离,GRN预测中提高0.03的AUROC;(4) 建立SCBENCH基准测试,系统性地评估LLM在组学原生推理方面的能力。
与已有工作不同的是,本文的独特切入角度在于提出了组学原生推理(Omics-Native Reasoning, ONR)这一全新范式。与现有方法的本质区别在于:(1) 不同于嵌入模型将数据压缩为不透明向量,SCPILOT要求LLM以自然语言明确阐述每个生物学推理步骤;(2) 不同于传统工具代理仅包装默认启发式方法,SCPILOT要求模型解释Monocle或SCENIC的输出并进行深入的生物学推理以发现新知识;(3) 不同于一次性提示(direct prompting),SCPILOT实现了迭代式的假设-验证-修正循环,模型可以在后续推理中修正先前的错误。具体而言,SCPILOT通过问题到文本的转换器将高维数据转化为语义摘要,同时保留生物学关键信息(如聚类大小、顶级标记基因、发育轨迹连接等),使LLM能够在有限上下文窗口内进行有意义的推理。
核心方法
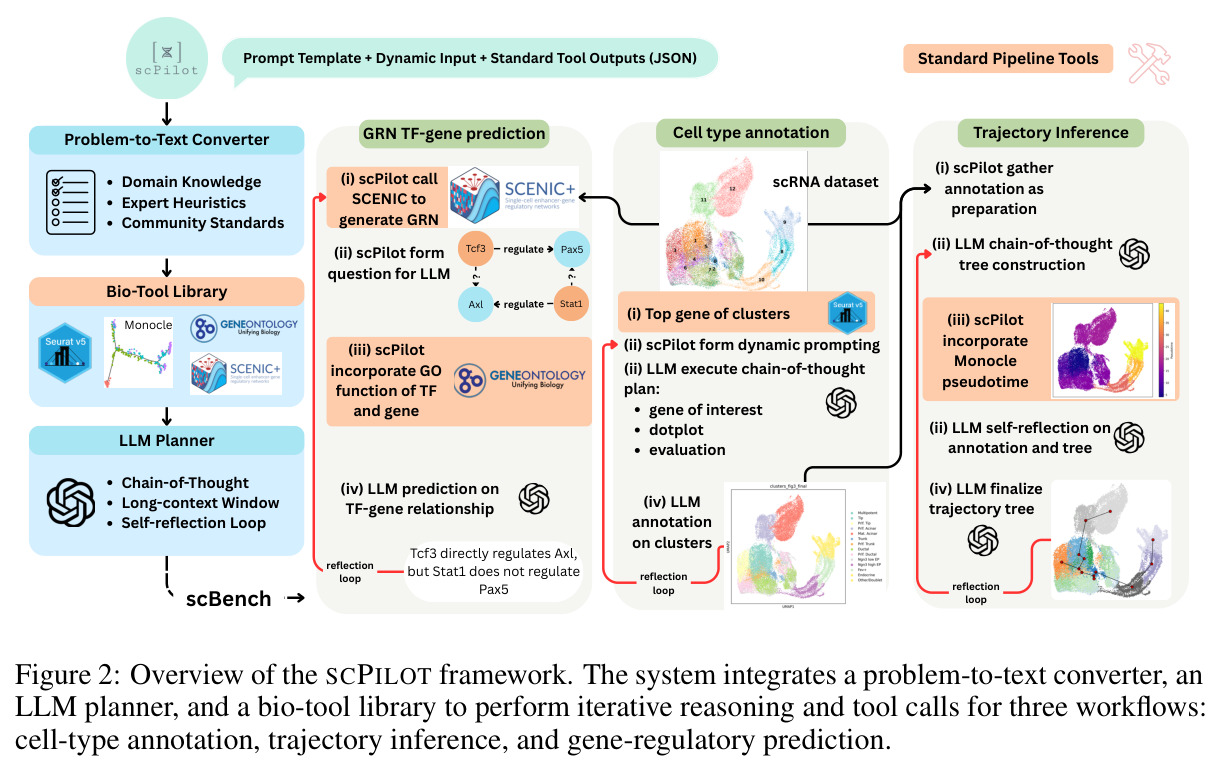
SCPILOT的整体思路可以类比为一个经验丰富的单细胞生物学家的分析流程:首先将复杂的高维数据转化为可理解的摘要信息,然后基于领域知识提出假设,接着调用专业工具获取证据,最后根据证据迭代修正结论。框架由三个核心组件构成:(1) 问题到文本转换器(Problem-to-Text Converter),将scRNA-seq表达矩阵转化为LLM可消化的文本摘要;(2) 生物工具库(Bio-Tool Library),封装了Scanpy、Monocle 3、pySCENIC等经典生物信息学工具作为可调用的原语算子;(3) LLM推理器(LLM Reasoner),基于强大的LLM(如o1或Gemini-2.5-Pro)进行链式推理和自我反思。整个推理流程可以形式化表示为:X→Prompt→{(Thought_k, Call_k)}_{k=1}^K→R_{1:K}→ŷ,其中模型在每一步k产生自然语言声明c_k和原语算子调用o_k,最终形成可审计的推理轨迹。
SCPILOT的核心创新在于组学原生推理(ONR)范式,其本质区别在于要求LLM直接在组学数据空间中进行推理,而非简单地包装工具输出。具体来说,ONR定义了一个逐步推理过程:设S_0 = X为初始数据状态,在每一步k,推理器产生一对(c_k, o_k),其中c_k是自然语言声明、论证或决策,o_k是应用于当前数据状态S_{k-1}的原语算子(如过滤、聚类、评分等),使得S_k = o_k(S_{k-1})。推理序列R = (c_1, o_1), ..., (c_K, o_K)构成了一个口头+计算的证明,最终状态S_K通过映射函数产生预测结果。这一范式与传统方法的根本区别在于:(1) 嵌入模型在向量空间中操作,无法提供语言解释;(2) 工具代理生成代码并执行,但推理隐藏在代码注释中,数值证据和生物学声明之间的因果逻辑链容易丢失;(3) ONR则要求每一步推理都同时包含自然语言论证和计算证据,形成完整的、可审计的推理轨迹。
方法步骤详情
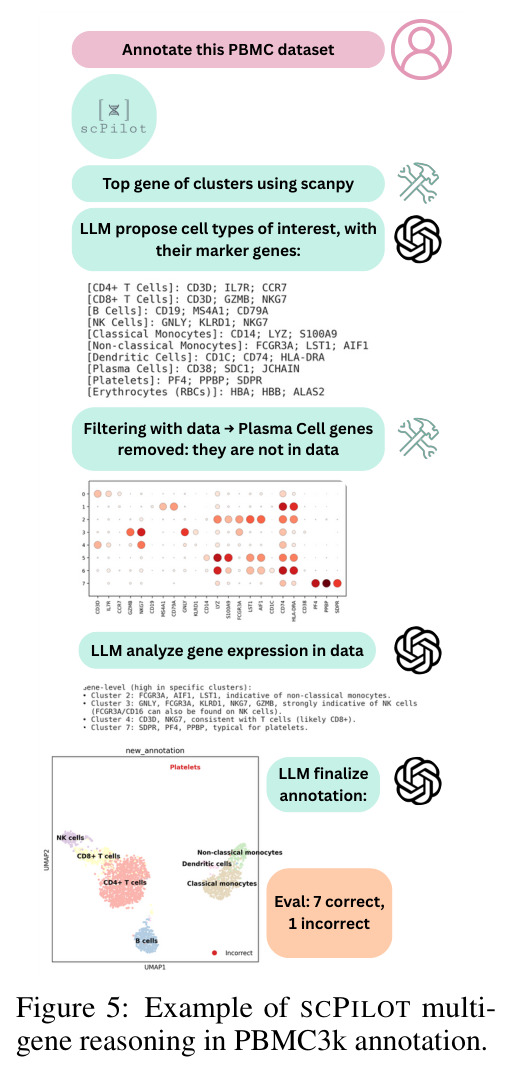
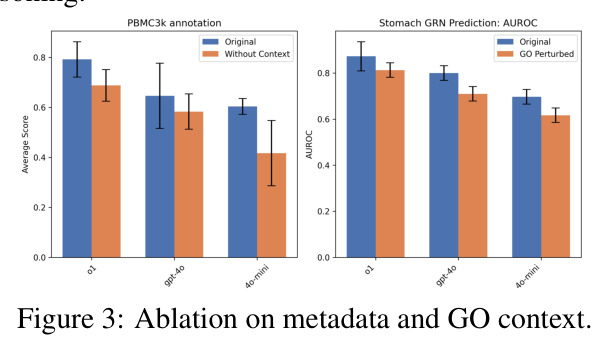
SCPILOT的方法步骤针对三个任务各有不同,但遵循统一的框架原则。对于细胞类型注释,步骤如下:(i) 问题到文本转换器使用Scanpy的Leiden聚类算法对细胞进行聚类,并提取每个聚类的前k=10个标记基因,形成文本摘要;(ii) LLM基于标记基因和数据集上下文信息(物种、组织类型、实验条件)提出候选细胞类型和对应的标记基因组合;(iii) 模型通过调用dotplot工具直接查看基因在原始数据中的表达模式,过滤掉不在数据中表达的基因(如排除浆细胞因为SDC1缺失);(iv) 模型通过迭代推理(最多3轮)修正注释,使用多基因逻辑解决标记基因歧义。对于轨迹推断,步骤为:(i) 使用PAGA或Monocle-3的谱系图和伪时间信息作为输入;(ii) LLM进行初始轨迹构建;(iii) 利用Monocle的诊断输出进行自我审计,修正树根、恢复经典肝系序列、进行层次调整。对于GRN预测,步骤为:(i) 从pySCENIC获取前M≤150个有基序支持的TF-基因对;(ii) 模型结合Gene Ontology功能注释和组织特异性表达上下文进行推理;(iii) 过滤虚假的GO重叠,整合表达上下文与已知调控通路信息。
技术新颖性
SCPILOT的技术新颖性体现在多个层面。首先,推理范式的创新:ONR是首个将LLM推理直接锚定在原始组学数据上的系统性框架,与先前的工作(如GPT-4用于细胞类型注释的单一任务)不同,SCPILOT将这一范式扩展到整个分析工作流。其次,数据压缩的创新:问题到文本转换器是算法性的(非学习的),针对不同任务设计了不同的压缩策略——细胞类型注释使用聚类标记基因,轨迹推断使用谱系图和伪时间,GRN预测使用TF-基因对和基序支持,这确保了在显著降低数据维度的同时保留关键生物学上下文。第三,评估体系的创新:SCBENCH是首个系统性评估组学原生推理能力的基准测试,包含9个专家策划的数据集和自动评估器,覆盖数值准确性和生物学推理有效性两个维度。最后,设计原则的系统化:SCPILOT遵循三个严格验证的设计原则——(a) 生物学上下文优先(提示始终包含关键生物学元数据),(b) 迭代推理(系统性地基于累积证据修正假设),(c) 最小化人工启发式(性能提升仅通过增强提示策略实现)。
实验结果
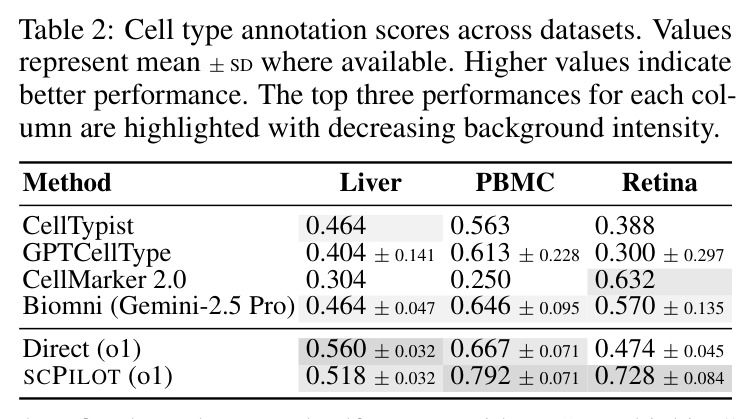
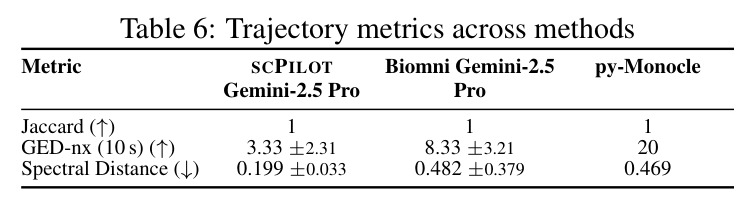
论文在8个模型(7个专有模型和1个开源模型)和9个数据集上进行了全面实验,得出以下核心发现。在细胞类型注释方面,SCPILOT在24个模型-数据集组合中的19个上优于直接提示方法。Retina数据集的中位准确率提升最大(+0.180),其次是PBMC3k(+0.042)和Liver(+0.024)。SCPILOT (o1)在PBMC3k上达到0.792,在Retina上达到0.728,在Liver上达到0.518。与传统工具相比,CellTypist仅达到0.464(Liver)、0.563(PBMC)、0.388(Retina),SCPILOT全面超越。在轨迹推断方面,SCPILOT在21个模型-指标对中的10个上进一步降低了结构误差,中位改进为GED降低2.0,谱距降低0.14。Gemini-2.5-Pro表现最优,在所有三个数据集上达到完美或接近完美的Jaccard分数(1.000),GED-nx仅为3.33(Liver)和9.50(Neocortex),相比Biomni的8.33和py-Monocle的20分别降低60%和52.5%。在GRN预测方面,SCPILOT (o1)在Stomach数据集上达到0.873的AUROC,相比直接提示的0.827提升+0.046。GPT-4o展现出最大的相对改进(平均AUROC +0.162),凸显了迭代推理在挖掘潜在调控洞察方面的有效性。三个一致的跨任务趋势是:(1) 大规模模型与结构化迭代推理的结合产生最优结果;(2) 迷你或延迟优化变体在扩展推理链中经常产生不可靠输出;(3) 迭代反思式提示显著提升了LLM在注释、轨迹推断和GRN预测中超越传统生物信息学方法的能力。

查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| 细胞类型注释(PBMC3k) | Cluster-level accuracy | SCPILOT (o1): 0.792 | 直接提示 (o1): 0.667; CellTypist: 0.563; GPTCellType: 0.613 | 相比直接提示提升 +0.125 (+18.7%),相比CellTypist提升 +0.229 (+40.7%) |
| 细胞类型注释(Retina) | Cluster-level accuracy | SCPILOT (o1): 0.728; SCPILOT (Gemini-2.0-Pro): 0.763 | 直接提示 (o1): 0.474; CellTypist: 0.388; CellMarker 2.0: 0.632 | SCPILOT (o1) 相比直接提示提升 +0.254 (+53.6%),为所有数据集中改进最大 |
| 细胞类型注释(Liver) | Cluster-level accuracy | SCPILOT (o1): 0.518 | 直接提示 (o1): 0.560; CellTypist: 0.464; Biomni: 0.464 | 相比CellTypist提升 +0.054 (+11.6%),注意:该数据集上直接提示略优于SCPILOT (o1) |
| 轨迹推断(Liver, GED-nx) | Graph Edit Distance (↓) | SCPILOT (Gemini-2.5-Pro): 3.33 | Biomni: 8.33; py-Monocle: 20 | 相比Biomni降低60%,相比py-Monocle降低83.4% |
| 轨迹推断(Jaccard平均) | Node-Jaccard (↑) | SCPILOT (Gemini-2.5-Pro): 1.000 | py-Monocle: 0.638-0.944 | 在所有数据集上达到完美分数 |
| GRN预测(Stomach) | AUROC | SCPILOT (o1): 0.873 | 直接提示 (o1): 0.827; GCN: 0.723; LLM4GRN: 0.727; BioGPT: 0.660 | 相比直接提示提升 +0.046 (+5.6%),相比GCN提升 +0.150 (+20.7%) |
| GRN预测(平均跨组织) | AUROC | SCPILOT平均AUROC提升 +0.098 | 直接提示基线 | GPT-4o展现出最大相对改进,平均AUROC +0.162 |
局限与改进
论文承认的局限性包括以下几个方面:(1) 数据压缩的信息丢失:当前的文本压缩策略可能遗漏稀有细胞群体的微妙信号,因为压缩过程倾向于保留主要群体的标记基因,这对稀有细胞类型的检测构成了挑战。(2) 可扩展性问题:将ONR扩展到十亿级token上下文和集成检索增强推理链面临重大挑战,当前框架受限于LLM的上下文窗口大小。(3) 幻觉风险:确保可信度需要鲁棒的方法来缓解LLM幻觉和错误声明,模型可能生成看似合理但生物学上不正确的推理。(4) 湿实验验证缺失:论文未整合实验湿实验室反馈来验证计算预测,这是一个关键的未来方向。(5) 开源模型的局限性:Gemma-3-27B在所有任务上显著落后于专有模型(如在PBMC3k上仅达到0.479 vs. o1的0.667),且推理速度慢15倍(135.7秒 vs. 8.8秒),使得完全本地部署在经济和操作上对大多数实验室不切实际。此外,作者还观察到在少数情况下,更简单的直接提示方法反而优于SCPILOT,这些反直觉的结果值得深入分析。作者的补充材料中还指出,SCPILOT的性能高度依赖于预处理工具的质量(如Monocle的输入如果被破坏,推理准确率会下降),这表明框架的鲁棒性仍有提升空间。
独立分析的弱点
基于对论文的深入分析,我识别出以下几个关键弱点:(1) 评估范围有限:SCBENCH仅包含9个数据集,且集中在PBMC、肝脏、视网膜等有限组织类型上,缺乏对更多样化组织(如脑区亚区、肿瘤微环境)和跨物种评估的支持。改进方向是扩展基准测试到更多组织类型和物种,尤其是临床相关的疾病样本。(2) 推理迭代次数固定:SCPILOT为细胞类型注释固定了最多3次推理迭代,这可能在简单数据集上浪费计算资源,而在复杂数据集上又不够充分。改进方向是设计自适应停止策略,根据推理置信度动态调整迭代次数。(3) 工具依赖性强:框架的性能高度依赖于预处理工具(如Monocle、Scanpy)的质量,如消融实验所示,当Monocle输入被破坏时推理准确率显著下降。改进方向是增加对工具输出的异常检测和自动修正机制。(4) 缺乏多模态整合:当前框架仅处理基因表达数据,未整合蛋白质组、表观基因组或空间转录组信息,限制了其在多组学分析中的应用。改进方向是将ONR范式扩展到多模态组学数据。(5) 成本分析不充分:虽然论文提到每任务仅需几美分,但未详细分析不同模型在不同任务上的成本差异,也未与人工分析的时间成本进行对比。
未来方向
论文作者提出了以下未来研究方向:(1) 增强数据表示方法:开发更精细的数据压缩策略以捕获稀有细胞群体的微妙信号,可能通过自适应采样或多尺度压缩实现。(2) 扩展到更大上下文:将ONR扩展到十亿级token上下文,集成检索增强推理链(RAG),使模型能够参考更广泛的文献和数据库。(3) 缓解幻觉:开发鲁棒的方法来检测和缓解LLM在生物学推理中的幻觉,可能通过交叉验证机制或多模型共识实现。(4) 整合湿实验反馈:将计算预测与湿实验室验证形成闭环,确认ONR框架的生物学有效性。基于本文成果可延伸的方向包括:(5) 多组学ONR:将框架扩展到蛋白质组学、表观基因组学和空间转录组学,实现真正的多模态组学推理。(6) 临床应用:将SCPILOT应用于疾病诊断和药物靶点发现,如肿瘤异质性分析和免疫治疗响应预测。(7) 交互式科学发现:开发人机协作界面,让生物学家能够与SCPILOT进行实时对话,引导推理方向并验证中间结论。(8) 自适应推理策略:设计元学习框架,使模型能够根据任务复杂度自动选择最优的推理深度和工具组合。
复现评估
论文在可复现性方面做得相对完善。代码和数据开源:论文明确声明代码、数据和包均在 https://github.com/maitrix-org/scPilot 开源,这为社区复现提供了基础。数据集可获取性:SCBENCH使用的9个数据集均来自高质量的公开scRNA-seq研究(如10x Genomics的PBMC3k、已发表的肝脏和视网膜数据集),ground truth来自原始论文的专家标注或TRRUST v2数据库的实验验证边。计算资源要求:论文使用了多个API模型(GPT-4o、o1、Gemini系列),无需本地GPU即可复现主要结果;但对于开源模型Gemma-3的评估需要4块NVIDIA A100 (80GB) GPU,推理速度为135.7秒/评估,这对大多数实验室来说是显著的硬件门槛。成本可承受性:使用Gemini-2.5-Pro时,最复杂的任务仅需几美分,这使得大规模复现实验在经济上可行。评估方法透明:论文提供了详细的评估指标(包括Jaccard、GED-nx、谱距、AUROC等)和消融实验设置。潜在挑战:复现的主要挑战在于API模型版本的时效性(论文评估的模型版本可能随时间更新)、随机性(LLM输出的非确定性)以及预处理工具(如Scanpy、Monocle版本)的一致性。
论文图表
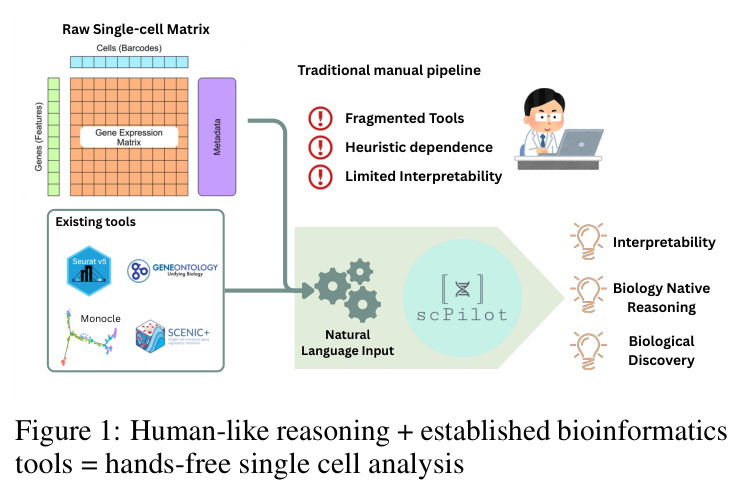
该图展示了SCPILOT的核心理念对比。左侧是传统的手动分析流程(Manual Pipeline),包括碎片化工具调用、启发式依赖和有限的可解释性;右侧展示了SCPILOT的Omics-Native Reasoning范式,将生物学原生推理、自然语言输入和现有工具(如Monocle)有机结合,实现可解释的生物学发现。图中用流程图形式展示了从原始单细胞矩阵到生物学发现的两条路径。
这张图是理解论文核心动机和创新点的关键,直观地展示了传统方法与ONR范式的根本区别,帮助读者快速把握论文的切入角度。