基于多尺度结构生成的蛋白质自回归建模 Protein Autoregressive Modeling via Multiscale Structure Generation
首个蛋白质骨架多尺度自回归生成框架PAR,实现从粗到精的结构生成
前置知识
自回归建模 (Autoregressive Modeling)
自回归模型是一类生成模型,它通过逐步预测序列中的下一个元素来建模联合概率分布。在语言模型中表现为 next-token prediction,在本文中扩展为 next-scale prediction。自回归模型的核心公式为 $p_\theta(x) = \prod_{i=1}^{n} p_\theta(x_i | x_{<i})$,即当前元素的生成条件依赖于所有之前的元素。自回归模型因其出色的可扩展性和零样本泛化能力在 NLP 和视觉领域取得了巨大成功,如 GPT 系列和 VAR 模型。
理解自回归建模是理解本文核心框架的前提,本文首次将自回归范式引入蛋白质结构生成领域。
Flow Matching (流匹配)
Flow Matching 是一种连续归一化流(CNF)的训练方法,通过定义从先验分布(如标准高斯噪声)到目标数据分布的线性插值路径来训练生成模型。给定噪声 $\epsilon \sim \mathcal{N}(0, I)$、时间变量 $t \in [0,1]$ 和目标数据 $x$,插值样本为 $x_t = t \cdot x + (1-t) \cdot \epsilon$,模型学习预测速度场 $v_\theta$ 来描述从噪声到数据的映射。相比扩散模型,Flow Matching 的训练目标更简洁,采样可以通过 ODE 或 SDE 实现。
PAR 使用 Flow Matching 作为其骨干解码器来直接建模 Cα 原子坐标,是方法架构的关键组成部分。
蛋白质骨架 (Protein Backbone)
蛋白质骨架是指蛋白质主链上的原子,主要包括 N、Cα、C 三个原子。在简化建模中,通常只考虑 Cα 原子的三维坐标,即 $x \in \mathbb{R}^{L \times 3}$,其中 $L$ 是残基数。蛋白质骨架决定了蛋白质的三维折叠结构和功能。结构生成任务的目标就是学习如何从随机噪声中生成合理的蛋白质骨架坐标。
本文的目标是生成蛋白质骨架结构,理解骨架的概念和表示方式是理解实验评估指标的基础。
暴露偏差 (Exposure Bias)
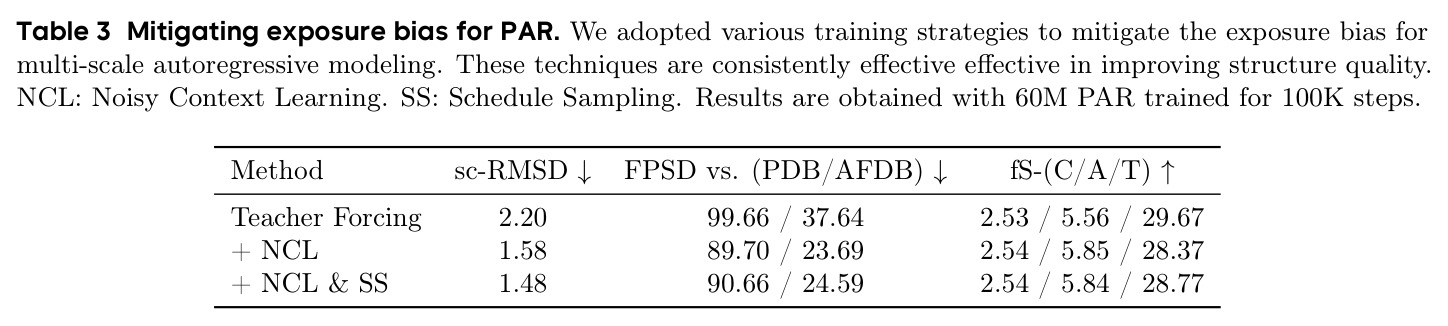
暴露偏差是自回归模型中的一个核心问题,源于训练时使用 teacher forcing(将真实数据作为条件输入)与推理时使用模型自身预测作为条件之间的不匹配。训练时模型从未见过自身的错误预测,但推理时必须基于自己的预测逐步生成,这会导致误差累积,严重降低生成质量。在本文的初步实验中,teacher forcing 大幅降低了生成结构的可设计性。
暴露偏差是自回归模型应用于蛋白质结构生成的主要障碍之一,本文提出的技术创新正是为了解决这一问题。
设计性 (Designability)
设计性是评估蛋白质结构生成质量的关键指标。评估流程为:首先用 ProteinMPNN 为生成的骨架结构反向设计氨基酸序列(生成 8 条候选序列),然后用 ESMFold 将序列折叠回三维结构,最后计算预测结构与原始生成结构之间的 Cα-RMSD。如果最小 RMSD 低于 2 Å,则认为该结构是可设计的。设计性越高说明生成的结构越合理、越接近真实的蛋白质结构。
设计性是本文最重要的评估指标之一,PAR 达到了 96.6% 的设计性,是衡量方法有效性的核心证据。
Fréchet Protein Structure Distance (FPSD)
FPSD 是类比图像生成中 FID(Fréchet Inception Distance)提出的蛋白质结构分布度量指标。它将蛋白质结构嵌入到折叠类别预测器定义的特征空间中,然后基于高斯近似计算生成分布与参考分布之间的 Wasserstein 距离。FPSD 越低表示生成的结构分布越接近真实蛋白质结构分布,能够同时反映生成质量和多样性。
FPSD 是本文评估模型捕获数据分布能力的主要指标,PAR 在 PDB 上达到了 161.0 的 FPSD 分数。
研究动机
蛋白质结构生成是生物设计领域的核心任务,现有方法主要依赖扩散模型和流匹配模型,如 FrameDiff、RFDiffusion、Proteina 等。这些方法虽然取得了一定成功,但都是单尺度模型,只能在单一粒度上操作,无法灵活处理不同精细度的结构信息。与此同时,自回归建模在语言模型(如 GPT)和视觉生成(如 VAR)中展现出了卓越的可扩展性和零样本泛化能力,但在蛋白质结构生成领域几乎未被探索。仅有 Gaujac 等人的工作尝试将因果 Transformer 应用于离散结构 token 的建模,但离散化会损失结构细节,且单向自回归顺序与蛋白质残基之间的双向空间依赖性存在根本冲突——序列上远距离的残基在三维空间中可能紧密接触形成氢键或疏水作用。
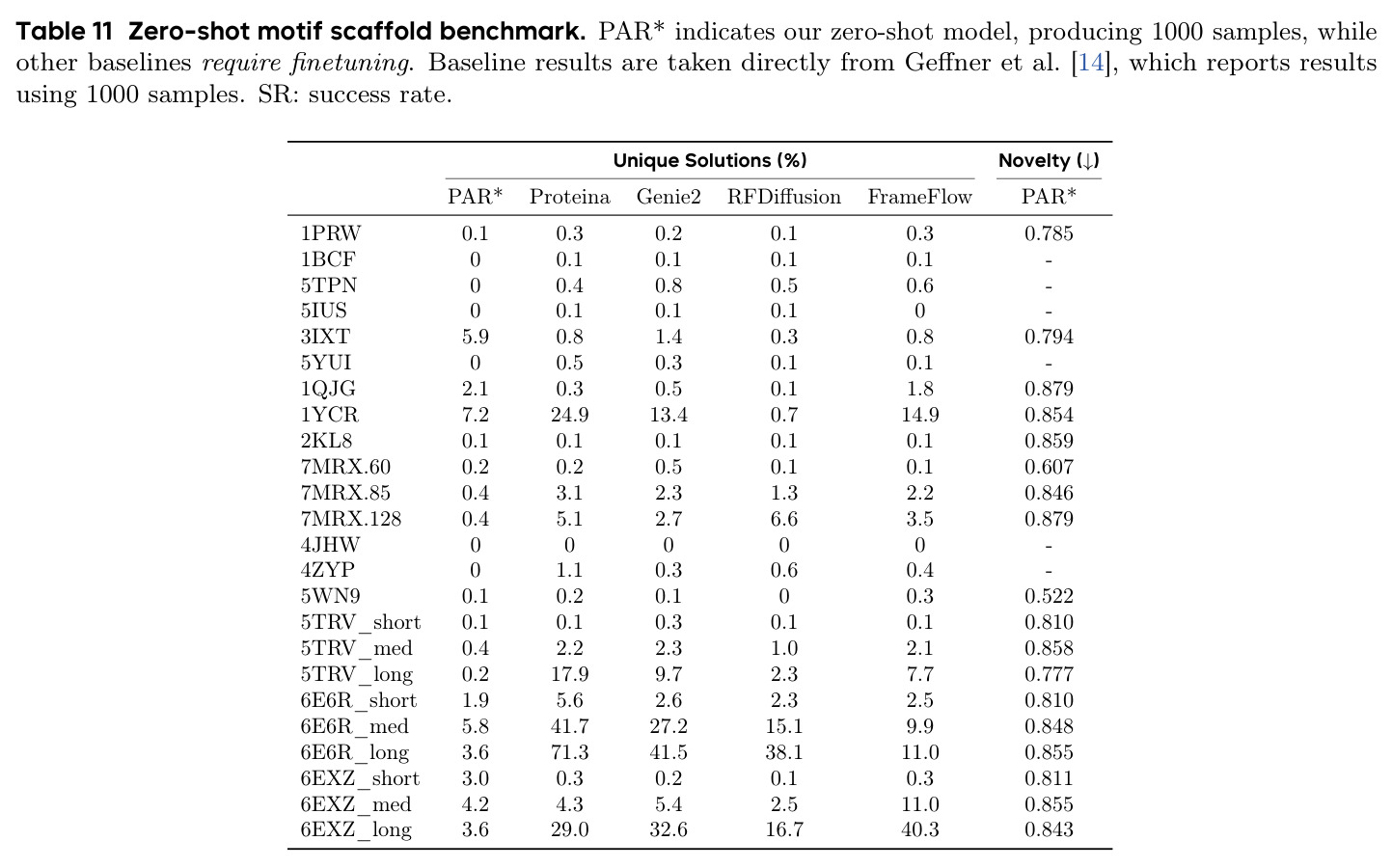
本文的目标是本文的核心目标是设计一个能够克服上述限制的蛋白质自回归框架。具体而言,作者希望:(1)避免离散化蛋白质结构,直接在连续空间中建模 Cα 原子坐标,保留结构细节;(2)解决蛋白质残基双向依赖与自回归单向假设之间的矛盾;(3)缓解自回归模型固有的暴露偏差问题;(4)利用自回归模型的特性实现零样本条件生成和 motif scaffolding,无需额外微调。
与已有工作不同的是,本文的独特切入角度是借鉴蛋白质结构的层次化本质。蛋白质结构天然具有多尺度特性,从粗粒度的全局拓扑和三级折叠,到中间的二级结构,再到最精细的原子坐标。作者提出了"next-scale prediction"的自回归策略,将传统的 next-token prediction 从序列维度扩展到尺度维度。这种设计灵感来源于图像生成中的 VAR 模型,但需要针对蛋白质结构的独特性质进行重新设计:(1)使用基于序列维度插值的多尺度下采样来构建结构层次;(2)使用 AR Transformer 产生尺度级条件嵌入来指导生成;(3)使用共享的 Flow Matching 骨干解码器在每个尺度上直接生成 Cα 坐标。这种多尺度自回归框架将蛋白质生成类比为"雕刻雕像"——先建立粗略的全局形态,再逐步精细化细节。
核心方法
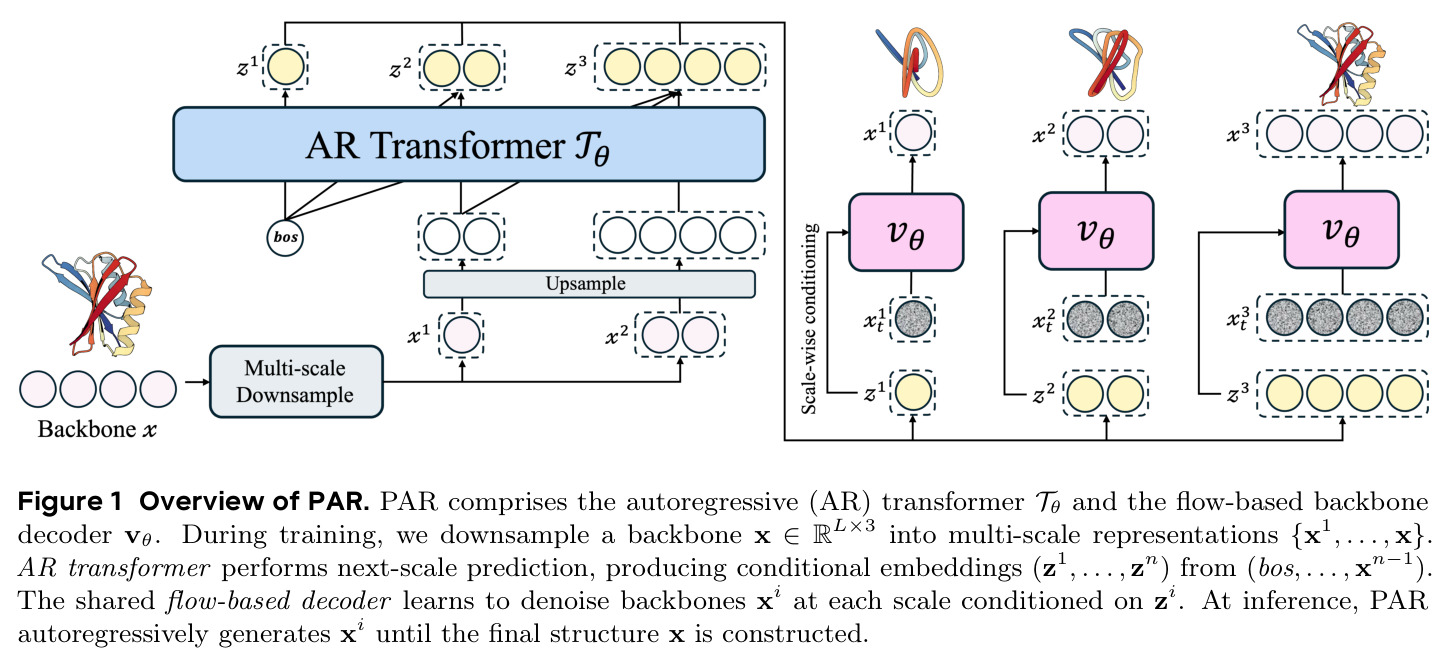
PAR(Protein AutoRegressive)是一个多尺度自回归框架,其核心直觉是:蛋白质结构天然具有层次性,从粗到细可以分解为多个尺度。就像雕刻一座雕像,先建立粗略的轮廓形态,再逐步添加细节。技术路线上,PAR 由三个核心组件组成:(1)多尺度下采样操作,将蛋白质骨架 $x \in \mathbb{R}^{L \times 3}$ 通过序列维度插值分解为多个尺度 $X = \{x_1, x_2, \ldots, x_n\}$;(2)自回归 Transformer $T_\theta$,编码所有前置尺度的信息,生成尺度级条件嵌入 $z_i$;(3)基于 Flow Matching 的骨干解码器 $v_\theta$,在条件嵌入的指导下直接生成 Cα 原子坐标。整体建模公式为 $p_\theta(x) = \prod_{i=1}^{n} p_\theta(x_i | z_i = T_\theta(X_{<i}))$,其中每个尺度的生成都条件依赖于所有更粗尺度的信息。
PAR 的核心创新在于"next-scale prediction"而非传统的"next-token prediction"。已有方法如 Gaujac 等人将蛋白质结构离散化为 token 序列进行自回归生成,这会导致结构细节损失且面临残基双向依赖与单向自回归的冲突。PAR 的本质区别在于:(1)它在连续空间中操作,使用 Flow Matching 解码器直接生成 Cα 坐标,避免了离散化损失;(2)它在尺度维度而非序列维度进行自回归,每个尺度内部保留了双向结构依赖,因为单个尺度内的所有残基是同时生成的而非逐步生成的;(3)使用 AR Transformer 产生条件嵌入来指导共享的 Flow Matching 解码器,而不是直接自回归生成坐标。这种设计使得 PAR 自然地包含 Flow Matching 模型作为单尺度特例,形成了更通用的框架。此外,通过 Noisy Context Learning 和 Scheduled Sampling 技术有效缓解了暴露偏差。
方法步骤详情
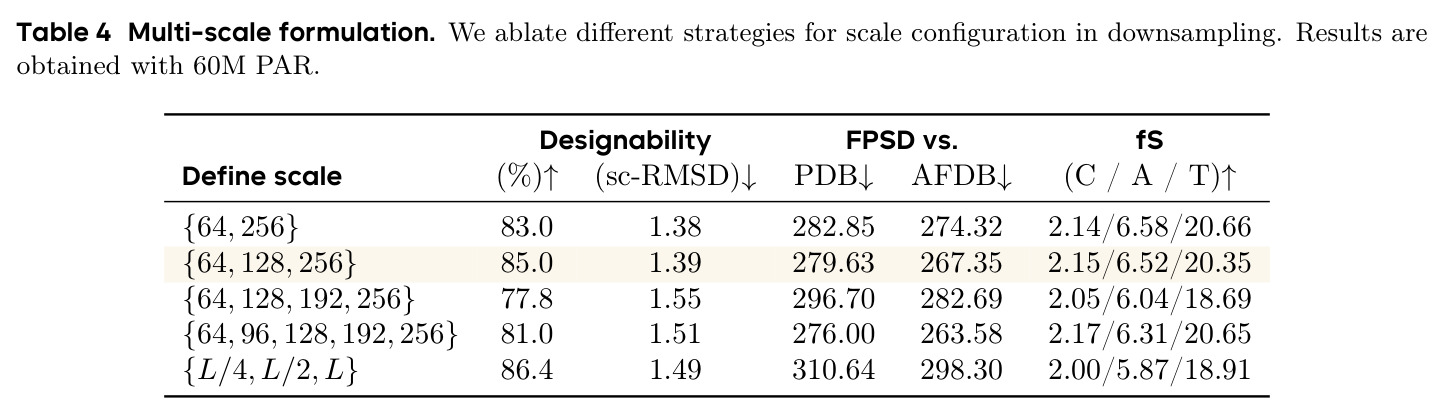
PAR 的完整方法流程包含以下步骤:(1)**多尺度下采样**:给定蛋白质骨架 $x \in \mathbb{R}^{L \times 3}$,通过序列维度插值将其分解为 $n$ 个尺度 $X = \{x_1, x_2, \ldots, x_n\}$,其中 $x_i = \text{Down}(x, \text{size}(i)) \in \mathbb{R}^{\text{size}(i) \times 3}$。默认配置为 $S = \{64, 128, 256\}$。(2)**自回归 Transformer 编码**:对于第 $i$ 个尺度,将前置尺度的结构拼接后输入 Transformer:$z_i = T_\theta(\text{bos}, \text{Up}(x_1, \text{size}(2)), \ldots, \text{Up}(x_{i-1}, \text{size}(i)))$,其中 bos 是可学习的起始嵌入,Up 是上采样插值操作。(3)**Flow Matching 解码**:使用条件嵌入 $z_i$ 通过自适应层归一化注入骨干解码器 $v_\theta$,在每个尺度上用 Flow Matching 损失训练:$\mathcal{L}(\theta) = \frac{1}{n} \sum_{i=1}^{n} \mathbb{E}[\|v_\theta(x_t^i, t_i, z_i) - (x^i - \epsilon_i)\|^2]$。(4)**缓解暴露偏差**:通过 Noisy Context Learning(给训练输入加噪声 $x_i^{\text{ncl}} = w_i \cdot x_i + (1-w_i) \cdot \epsilon_i$)和 Scheduled Sampling(以 0.5 概率用模型预测替换真实上下文)。(5)**推理**:从最粗尺度开始,AR Transformer 生成条件嵌入,Flow Matching 解码器生成结构,上采样后送回 Transformer 继续下一尺度,直到生成完整结构。采样可使用 ODE 或 SDE,通过 SDE/ODE 编排实现高效采样。
技术新颖性
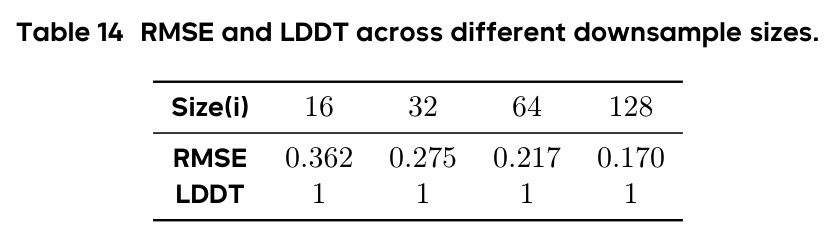
PAR 的技术新颖性体现在多个层面。首先,它是第一个将多尺度自回归建模应用于蛋白质骨架生成的工作,这一概念本身具有开创性。其次,在技术实现上,PAR 将自回归的维度从序列(token)扩展到了尺度(scale),这与 VAR 在图像生成中的思路一脉相承但需要针对蛋白质结构进行完全重新设计。具体创新包括:(1)使用序列维度插值(而非图像中的 2D 下采样)来构建多尺度表示,并证明这种 1D 下采样能很好地保持空间配对关系(RMSE 仅为 0.17-0.36,LDDT 始终为 1);(2)AR Transformer 不直接生成坐标,而是产生条件嵌入来指导 Flow Matching 解码器,结合了自回归模型的层次推理能力和流模型的连续空间建模能力;(3)Scale Embedding 和 Interpolated Position Embedding 两个技术细节帮助模型识别不同尺度和保持位置信息;(4)将 Noisy Context Learning 和 Scheduled Sampling 适配到多尺度蛋白质生成场景,有效缓解暴露偏差。理论上,PAR 证明了 Flow Matching 模型是其单尺度特例,使其成为更通用的生成框架。
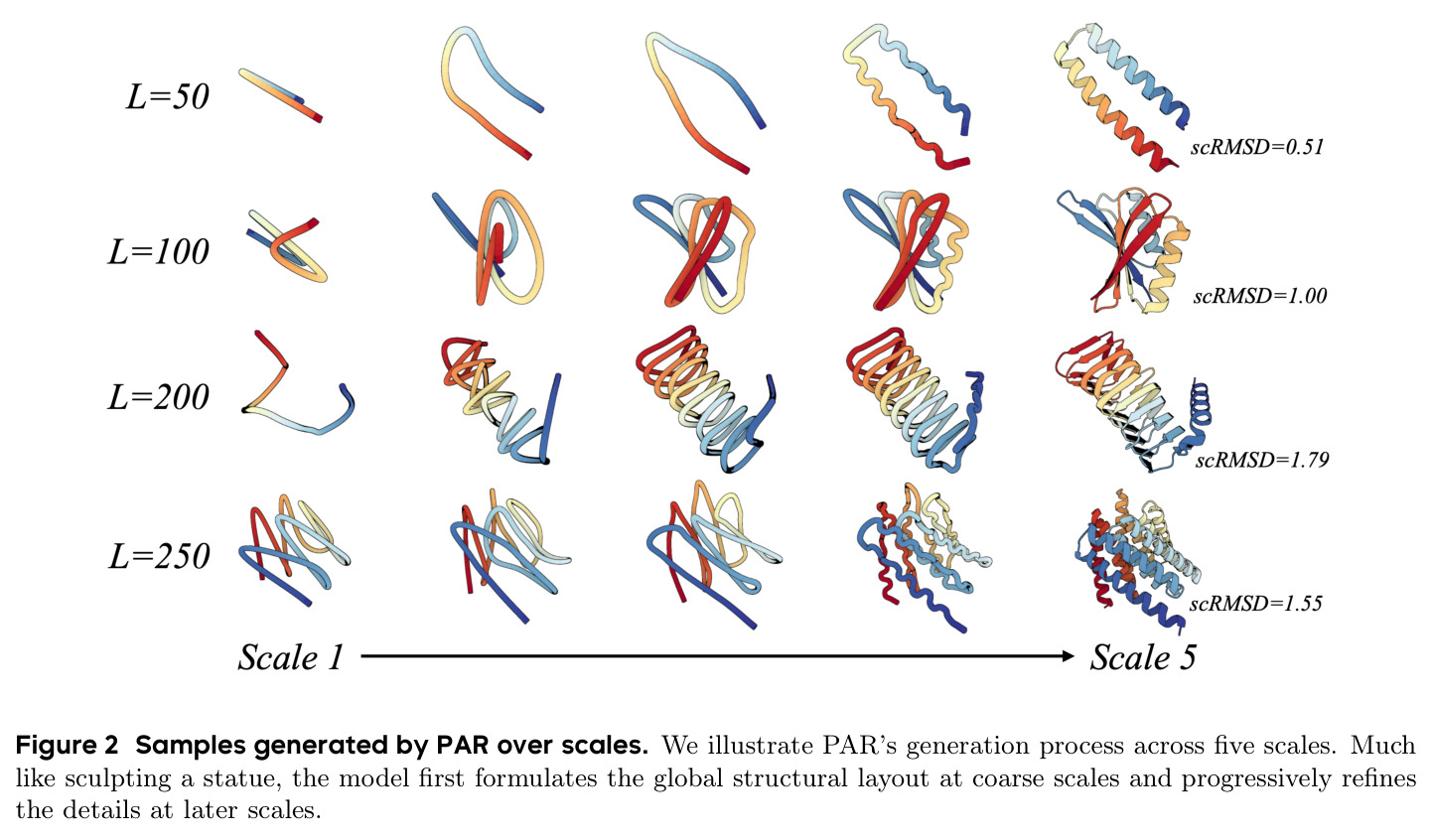
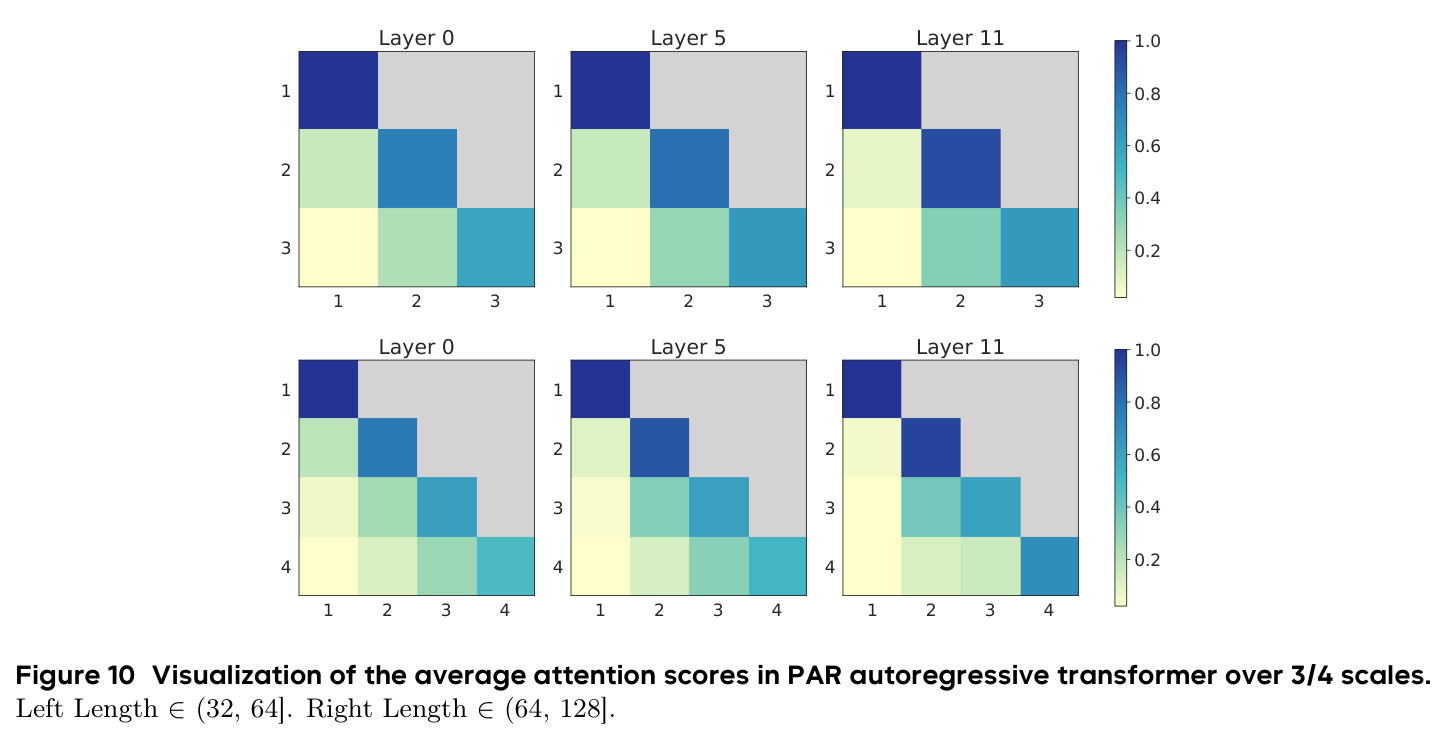
实验结果
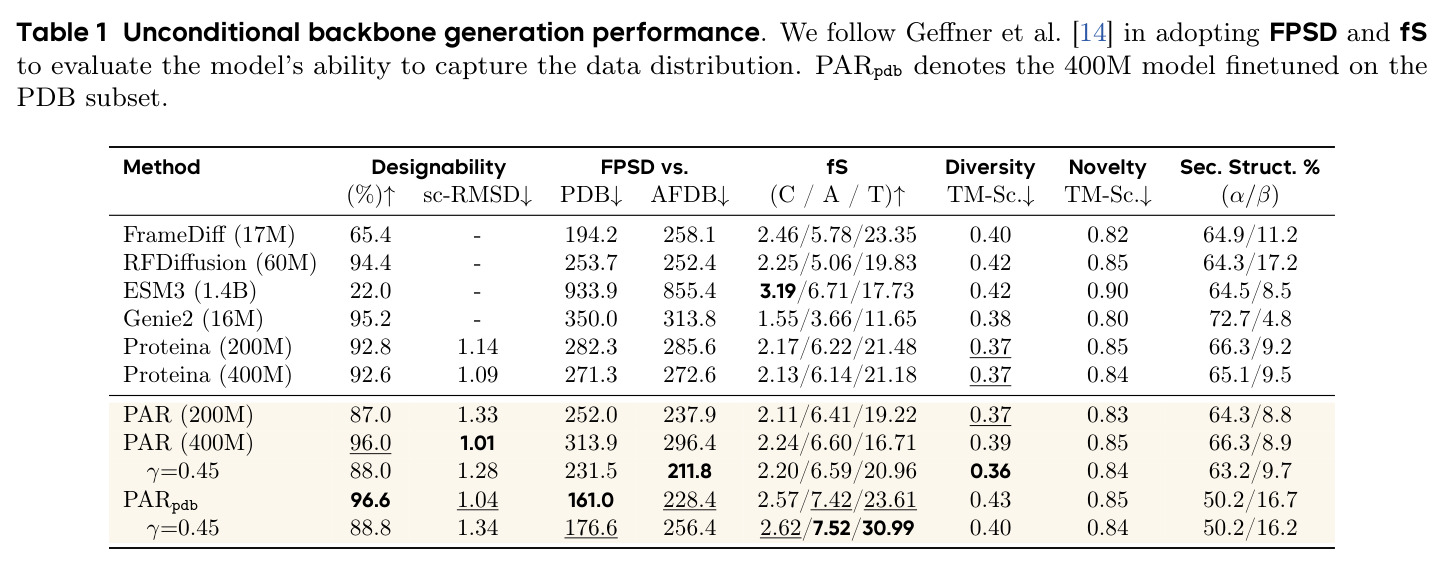
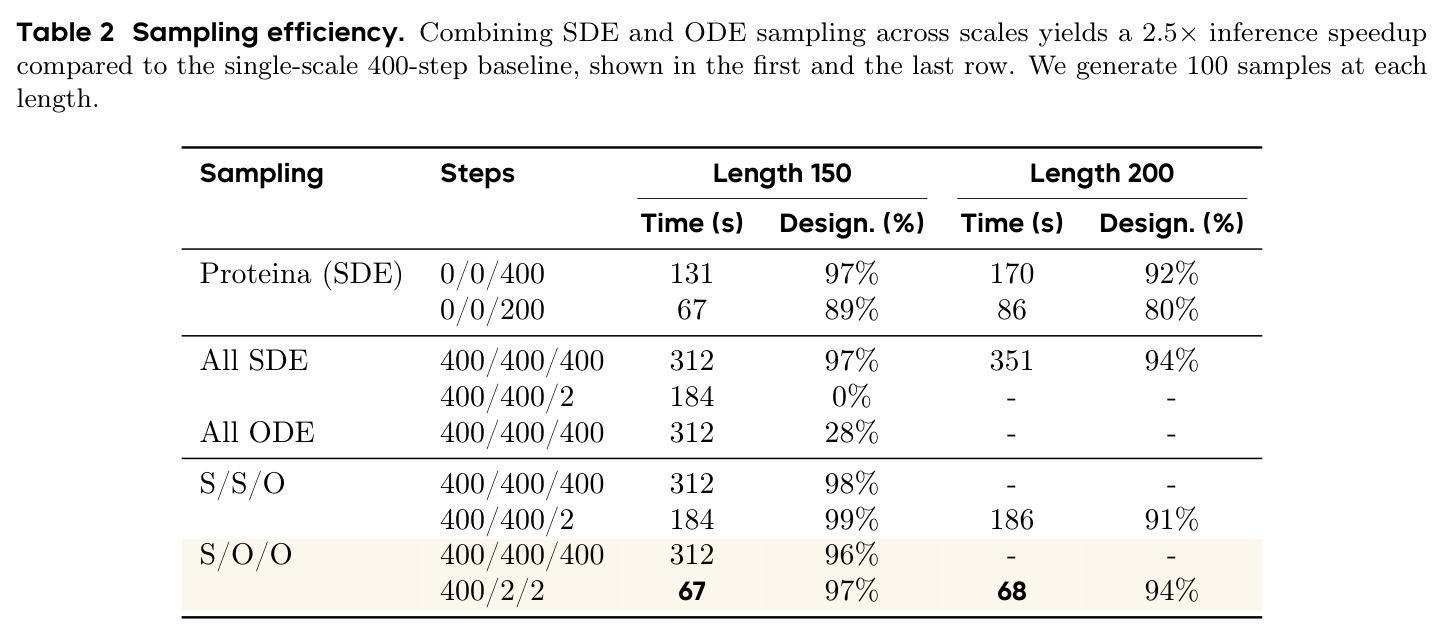
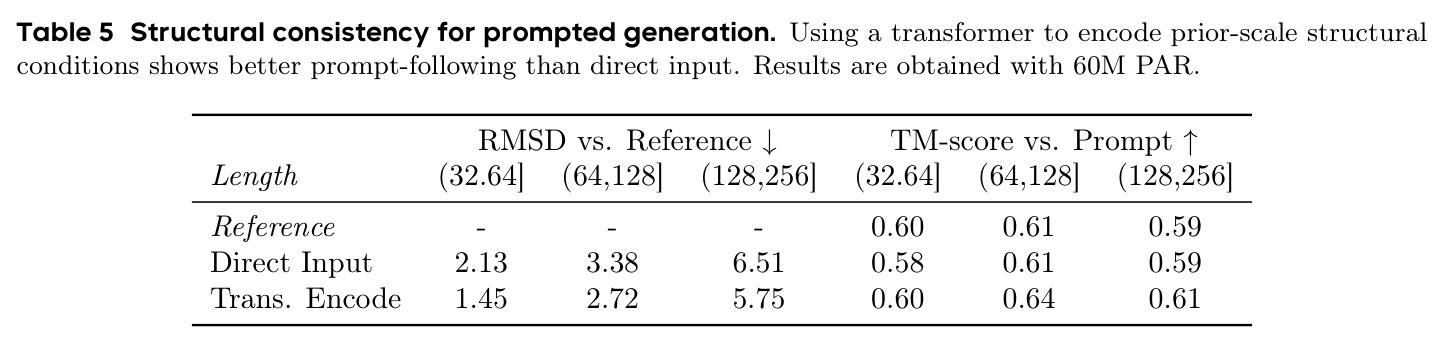
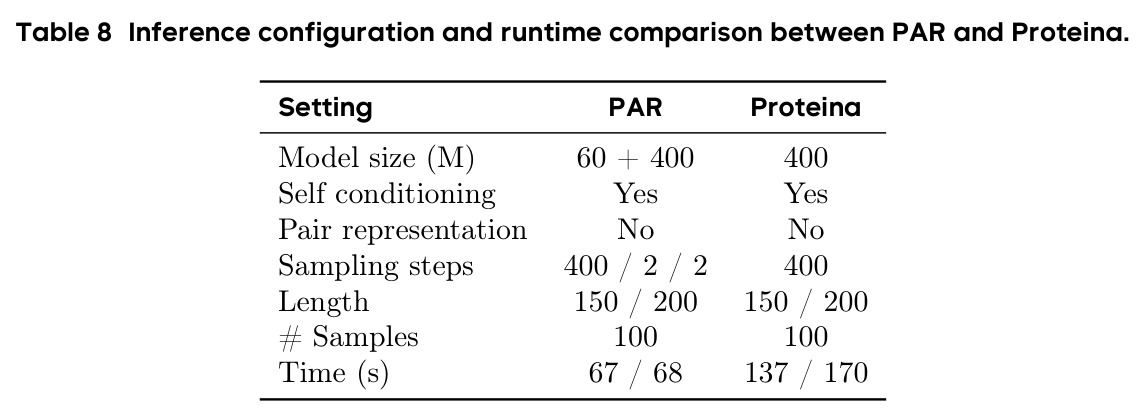
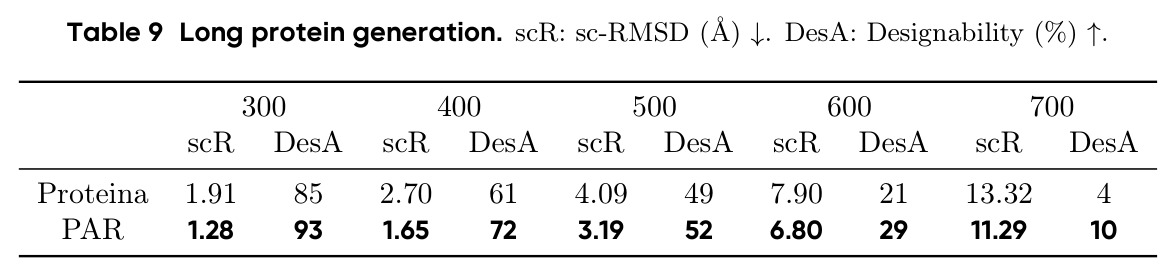
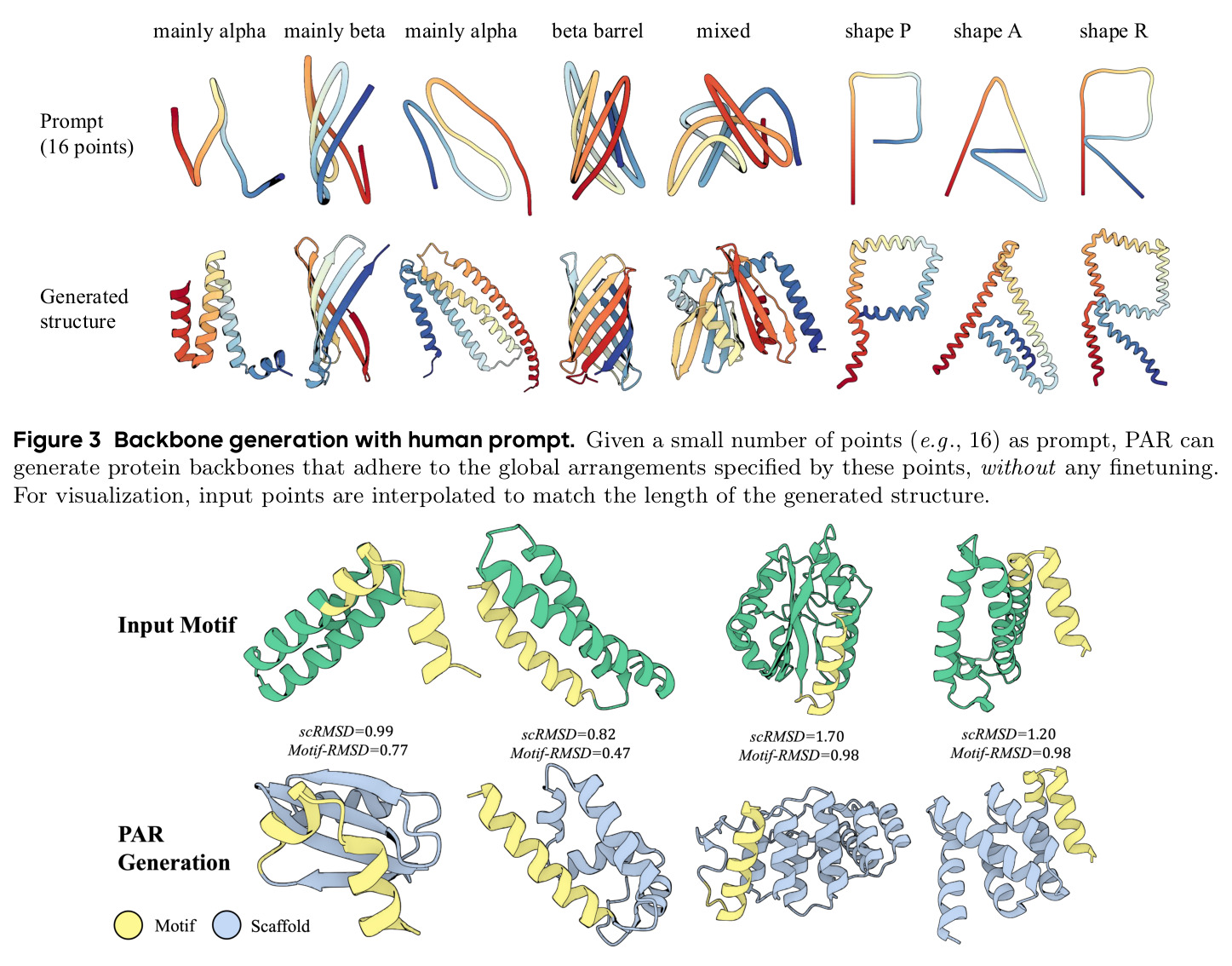
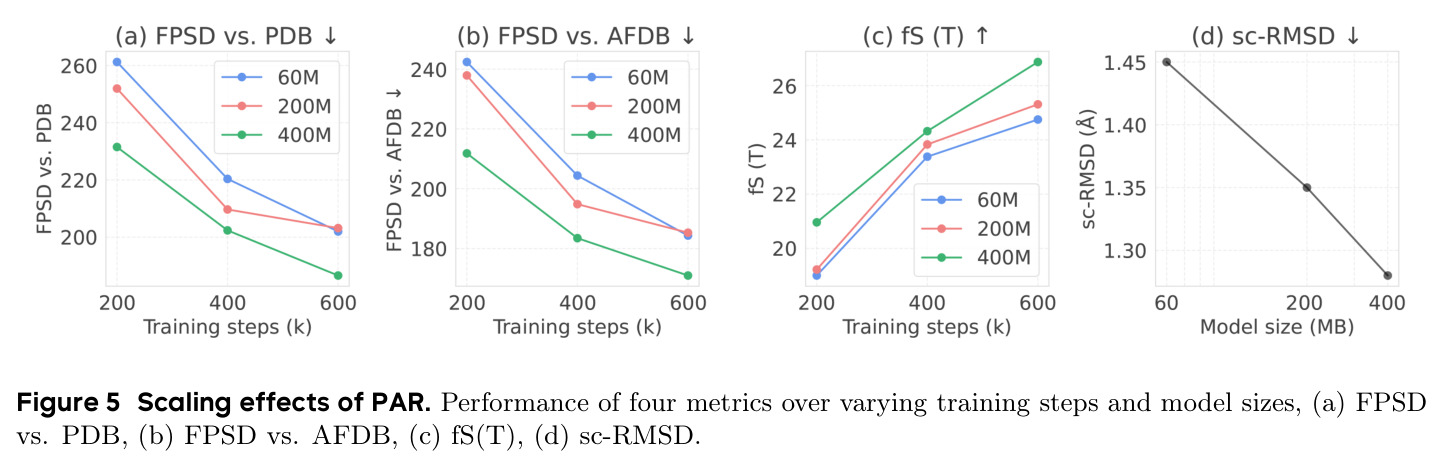
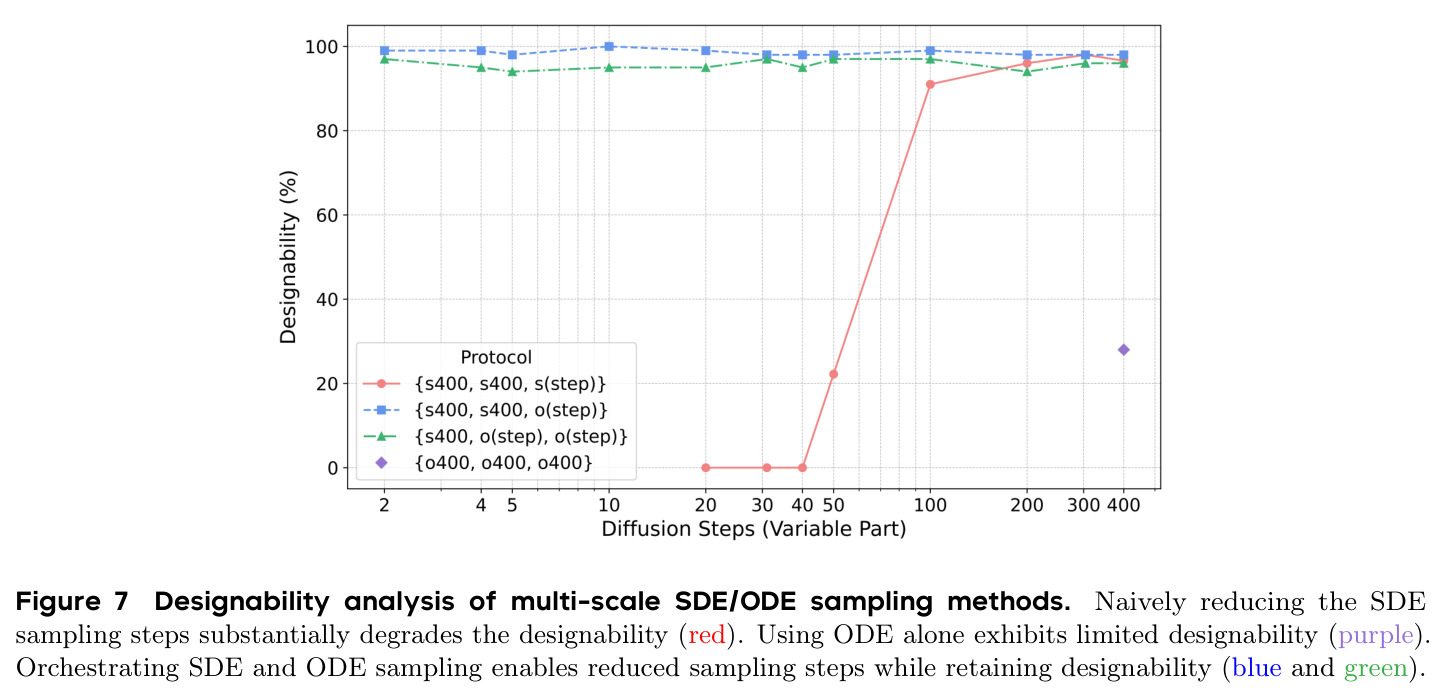
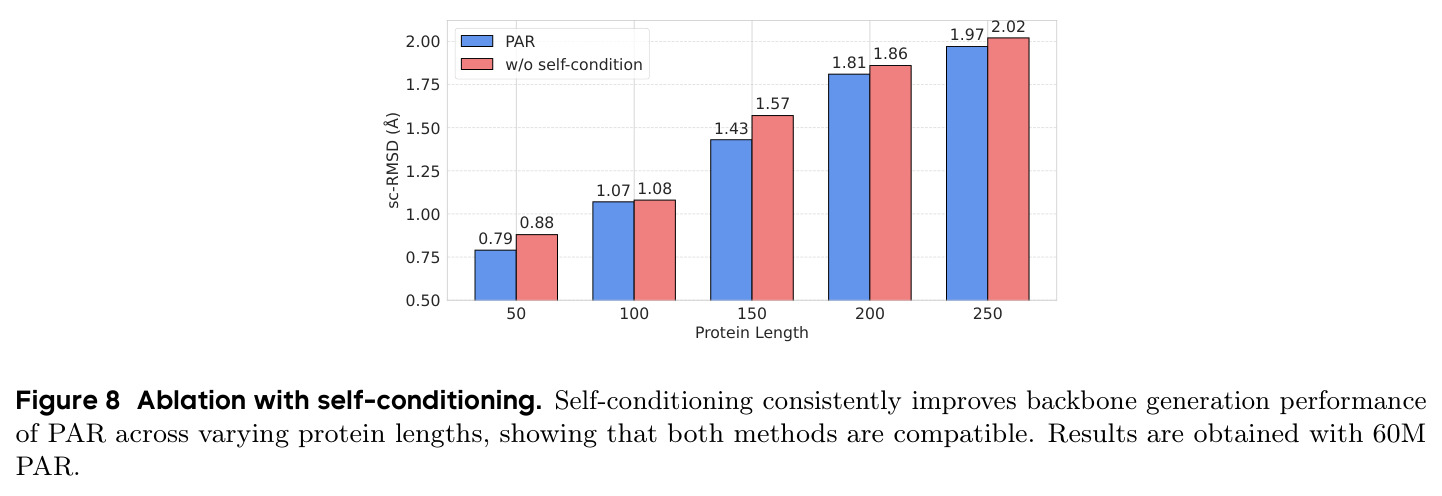
PAR 在多个维度上展示了出色的性能。在无条件生成基准测试中,使用 3 尺度配置 $S = \{64, 128, 256\}$ 的 PAR(400M 参数)达到了 96.0% 的设计性和 313.9 的 FPSD vs PDB 分数。经过 PDB 子集微调后,PARpdb 进一步提升至 96.6% 设计性和 161.0 FPSD vs PDB,显著优于所有基线方法,包括 Proteina(271.3)、RFDiffusion(253.7)和 FrameDiff(194.2)。在缩放效应实验中,PAR 展示了良好的缩放行为:增大模型规模从 60M 到 400M 可持续提升 FPSD 和 fS 分数,FPSD vs PDB 从 252 降至 187。在采样效率方面,通过多尺度 SDE/ODE 编排(第一尺度用 SDE 400 步,后续尺度用 ODE 2 步),PAR 实现了 2.5 倍采样加速,同时保持 97% 的设计性。在暴露偏差缓解方面,Noisy Context Learning 将 sc-RMSD 从 2.20 降至 1.58,FPSD vs AFDB 从 37.64 降至 23.69,加上 Scheduled Sampling 后进一步改善至 sc-RMSD 1.48。在零样本泛化方面,PAR 无需微调即可支持基于点提示的条件生成和 motif scaffolding,16 个点即可指定蛋白质的粗粒度形态。在长蛋白质生成方面,PAR 在 300-400 残基长度上优于 Proteina(设计性 93% vs 85% 和 72% vs 61%)。
查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| 无条件蛋白质骨架生成(PDB 微调后) | FPSD vs PDB | PARpdb: 161.0 | Proteina 400M: 271.3; RFDiffusion: 253.7; FrameDiff: 194.2 | 相比最强基线 FrameDiff 降低 17.2%,相比 Proteina 降低 40.6% |
| 无条件蛋白质骨架生成(PDB 微调后) | 设计性 (%) | PARpdb: 96.6% | Proteina 400M: 92.6%; RFDiffusion: 94.4%; Genie2: 95.2% | 超过所有基线,比次优 Genie2 提高 1.4 个百分点 |
| 无条件蛋白质骨架生成(AFDB) | FPSD vs AFDB | PAR 400M: 296.4 | Proteina 400M: 272.6; RFDiffusion: 252.4 | AFDB 基准上与基线竞争力相当 |
| 采样效率 | 推理时间(Length 200) | S/O/O 400/2/2: 68s | Proteina SDE 400步: 170s | 2.5 倍推理加速 |
| 暴露偏差缓解 | sc-RMSD (Å) | NCL + SS: 1.48 | Teacher Forcing: 2.20 | 降低 32.7% |
| 长蛋白质生成(Length 300) | 设计性 (%) | PAR: 93% | Proteina: 85% | 提高 8 个百分点 |
局限与改进
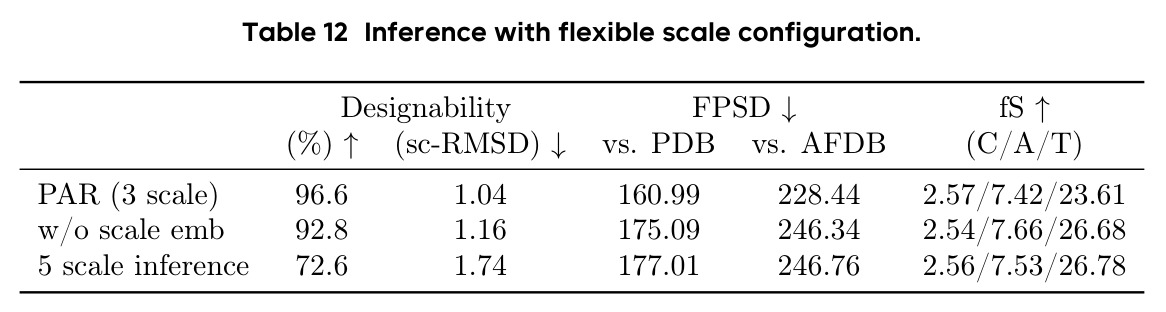
作者在论文中承认了几个重要局限性。首先,在 FPSD vs AFDB 指标上,PAR(296.4)并不优于 Proteina(272.6),说明在匹配 AFDB 分布方面仍有改进空间。其次,多尺度配置的选择是一个权衡:从 2 尺度增加到 3 尺度提升了性能,但增加到 4-5 尺度反而导致设计性下降(77.8%-81.0% vs 85.0%),可能是因为误差累积和暴露偏差在更多尺度上被放大。第三,长蛋白质生成仍然是挑战,在 500-700 残基长度上设计性显著下降(52%-10%),受限于训练数据的长尾分布。第四,PAR 只建模 Cα 原子,尚未扩展到全原子建模。第五,作者指出 Scale Embedding 的维度固定为训练时的尺度数,导致模型不能直接在不同的尺度配置下推理,虽然可以通过微调解决但设计性会下降(从 96.6% 降至 72.6%)。此外,从我的观察来看,论文缺乏与最新扩散模型(如 RFDiffusion2)的直接对比,且所有实验都限制在最长 256 残基的蛋白质上(长蛋白质实验需要额外微调),限制了对方法在真实应用场景中表现的全面评估。
独立分析的弱点
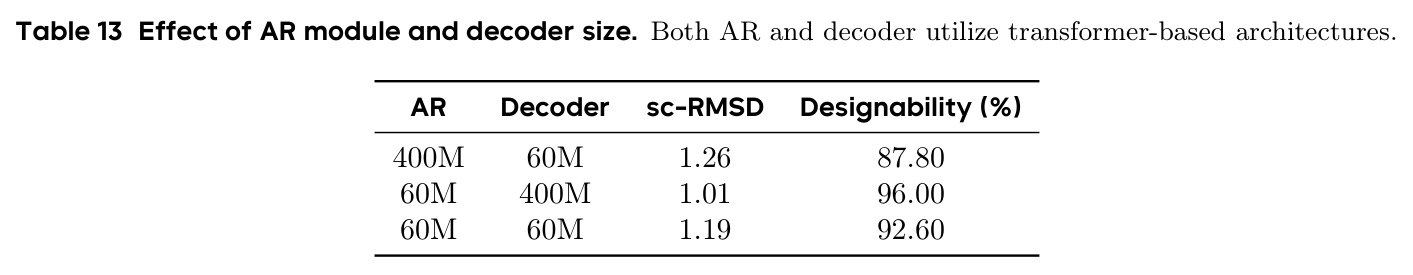
PAR 存在以下几个可以改进的弱点。首先,暴露偏差问题虽然通过 NCL 和 SS 得到缓解但未完全解决,作者自己也承认更大的 AR Transformer(400M)反而不如 60M 表现好,这说明暴露偏差在更大模型上更加严重。改进方向可以探索更先进的暴露偏差缓解技术,如利用强化学习或对抗训练来让模型更好地适应自身预测的分布。其次,多尺度下采样使用简单的线性插值,可能丢失蛋白质结构中的局部二级结构信息。可以考虑使用基于生物物理先验的下采样策略,如保持局部氢键网络或二级结构完整性。第三,尺度配置需要手动选择(默认 $S = \{64, 128, 256\}$),缺乏自适应确定最优尺度数量和大小的机制。第四,推理时 ODE 采样在所有尺度上仅有 28% 的设计性,说明模型对采样策略高度敏感,需要探索更鲁棒的采样方法。第五,论文仅评估了单链蛋白质,未考虑多链复合物或与配体的相互作用生成。
未来方向
作者提出了几个值得探索的未来方向。首先,构象动力学建模:PAR 可以零样本建模构象分布——将同一结构下采样后用 PAR 上采样来模拟局部分子动力学。其次,全原子建模:在当前 Cα 框架基础上增加两个更细尺度——骨架原子尺度(N, Cα, C)和全原子尺度(包括侧链),可以采用 P(all-atom) 风格设计或利用 Full-atom MPNN (FAMPNN) 从 PAR 生成的骨架预测全原子结构。第三,多链和其他生物分子模态:引入链标识符来区分不同生物分子并建模它们的联合结构;对于 DNA/RNA 等线性聚合物可以使用类似蛋白质的插值下采样,对于配体等非线性小分子则需要基于碎片的粗粒化策略。基于本文成果还可以延伸到:(1)利用 PAR 的多尺度特性进行蛋白质-蛋白质相互作用建模和 binder 设计;(2)将多尺度自回归框架应用于蛋白质序列和结构的联合生成;(3)探索更大规模的训练数据(目前 588K 结构远少于 ImageNet 的 1.28M)来缓解暴露偏差和提升长蛋白质生成能力。
复现评估
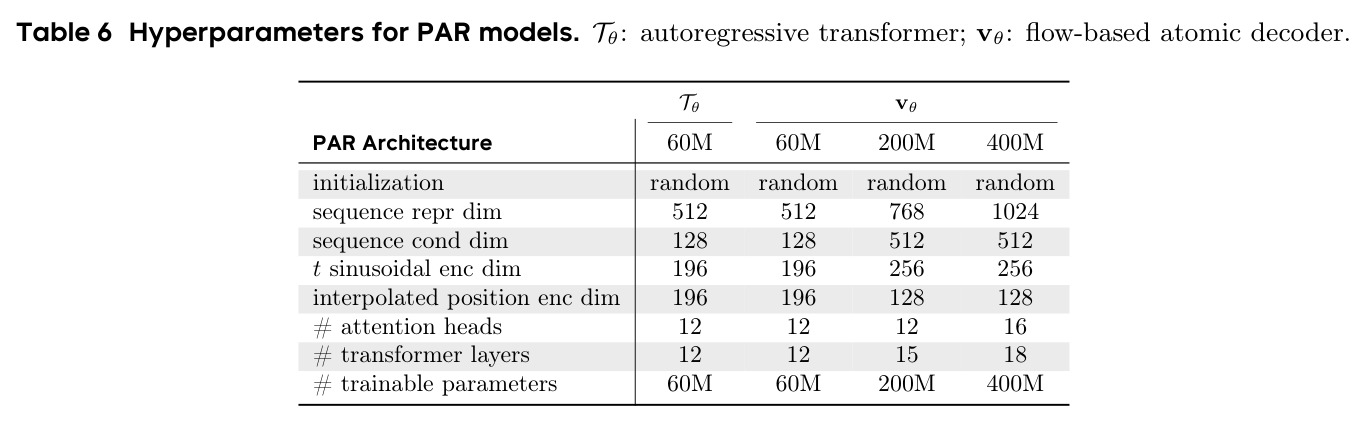
从复现角度来看,本文的实验设置相对清晰。训练使用 8 张 H100 GPU,batch size 每 GPU 为 15,训练 200K 步,100K 步的训练时间约为 23 小时(PAR 60+400M)。论文详细列出了模型架构超参数(Table 6),包括维度、层数、注意力头数等。然而,存在一些复现挑战:(1)论文基于 Proteina 的实现,但未明确说明是否开源代码;(2)PDB 微调数据集需要按照论文描述的过滤协议自行构建(21K 可设计样本),因为原始数据集不可公开获取;(3)评估依赖多个外部工具(ProteinMPNN、ESMFold、Foldseek)和预训练数据库,需要额外下载和配置;(4)AFDB 代表数据集的处理细节依赖于 Proteina 的预处理流程。总体而言,如果有足够的计算资源(8 张 H100)并遵循 Proteina 的训练流程,复现论文结果应该是可行的,但完整复现所有实验需要相当的时间和算力投入。
论文图表