M⁴olGen:精确多属性约束下的多智能体多阶段分子生成框架 M^4olGen: Multi-Agent, Multi-Stage Molecular Generation under Precise Multi-Property Constraints
两阶段框架:检索增强原型生成+GRPO片段优化,实现分子多属性精确控制
前置知识
分子属性约束生成
在药物发现和材料设计中,需要生成同时满足多个物理化学属性精确数值目标的分子。常见属性包括药物相似性(QED,取值0-1)、脂溶性(LogP,表征分子亲脂性)、分子量(MW,单位Da)以及前线轨道能级(HOMO/LUMO,单位eV)。传统方法通常优化单个或少数属性的代理函数,难以同时精确控制多个属性到指定数值范围。
本文的核心任务就是在多属性数值约束下生成分子,理解属性的物理含义和数值范围是理解实验结果的基础
BRICS片段分解
BRICS(Break Retrosynthetically Interesting Chemical Substructures)是一种基于逆合成分析的分子片段化方法,它在化学合成中实际形成或断裂的键处切割分子,生成的片段具有合成可及性和化学意义。每个分子可分解为片段集合,记为Phi(m) = {f_1, ..., f_k},并记录片段间的连接关系,形成可操作的编辑空间。
M⁴olGen的编辑操作(添加、删除、替换)都在BRICS片段层面进行,这是方法的核心操作单元
GRPO(Group Relative Policy Optimization)
GRPO是一种强化学习策略优化方法,最初由DeepSeek团队提出用于LLM的偏好学习和RLHF。其核心思想是对同一输入采样一组候选输出,根据奖励函数对候选进行排名,然后通过组内相对排名信号更新策略,使高排名候选的概率增加、低排名候选的概率降低。这种基于排名的更新方式对噪声奖励具有鲁棒性,无需ground-truth演示。
本文首次将GRPO应用于分子生成领域的数值条件优化,是方法的技术核心
检索增强生成(RAG)
检索增强生成是一种结合外部知识库检索与生成模型的技术范式。在本文中,给定目标属性向量p_tgt,系统从大规模分子语料库Omega中检索属性值在目标容差范围epsilon_i内的参考分子集合M,作为生成过程的锚点和示例,帮助模型在可行区域内进行编辑。
Stage I的原型生成依赖检索到的参考分子来引导片段编辑,是实现精确属性控制的关键设计
多跳优化(Multi-hop Refinement)
多跳优化是一种迭代精炼机制,允许模型在多个连续步骤中对分子进行逐步修改。每次hop执行一次片段级编辑操作,hop预算H控制结构复杂度和与原型的偏差。通过链式单跳编辑,模型可以累积小的局部改进,逐步逼近目标属性值,同时通过正则化器约束编辑复杂度。
实验表明从1-hop到3-hop性能单调提升,多跳机制是实现精确属性对齐的核心技术
研究动机
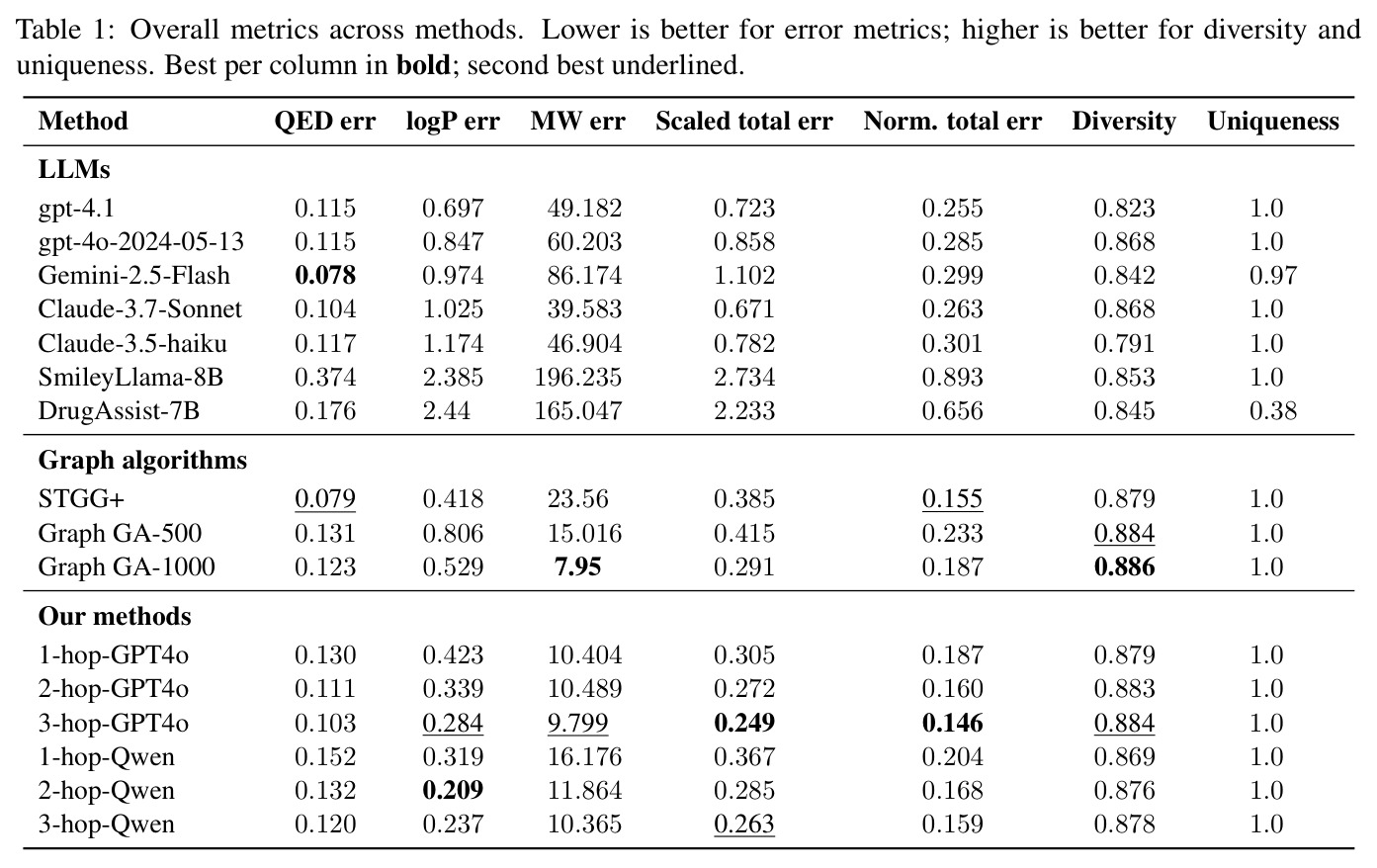
在药物开发、材料设计和分子从头设计等领域,生成满足精确数值属性约束的分子是一个关键且具有挑战性的任务。现有方法面临三大核心问题:第一,深度生成模型(如VAE、GAN、GCPN、GraphAF等)虽然在分子生成方面取得了进展,但多为单智能体方法,在精确满足多个数值约束方面存在探索-利用权衡的困难。第二,大型语言模型(如ChemGPT、ChemBERTa、MolT5等)虽然具有强大的表达能力,能够捕获化学语法和语义,但在精确数值推理和多属性控制方面表现不足,缺乏显式最小化目标距离的机制。第三,强化学习方法(如MolDQN、GFlowNets)支持属性驱动优化,但在多目标精确约束场景下难以平衡多个数值目标。实验数据显示,即使是最强的商业LLM(GPT-4.1)在QED、LogP、MW三属性约束下的归一化总误差仍高达0.255,而最强的图算法STGG+为0.155,仍有显著改进空间。
本文的目标是本文旨在开发一个能够在多个物理化学属性上实现精确数值控制的分子生成框架M⁴olGen。具体目标包括:第一,同时精确控制QED(药物相似性,0-1范围)、LogP(脂溶性)和MW(分子量)三个基本属性,以及HOMO和LUMO(前线轨道能级)两个能量属性;第二,构建一个大规模的、带有推理链的片段编辑数据集,支持可控的多跳推理训练;第三,验证多智能体检索增强原型生成与GRPO片段优化相结合的两阶段设计的有效性;第四,实现无需针对每个目标重新训练的泛化能力,并支持可控的编辑复杂度调节。
与已有工作不同的是,本文的独特切入角度体现在三个层面。首先,与现有LLM方法不同,M⁴olGen将分子生成问题形式化为在可操作片段编辑空间上的距离-目标优化问题,使用快速可验证的化学预言机(RDKit)提供精确的属性反馈,而非依赖LLM的数值推理能力。其次,与现有强化学习方法不同,本文首次将GRPO引入分子生成的数值条件优化场景,利用其组内相对排名的更新机制实现稳定且样本高效的策略优化。最后,与单阶段方法不同,本文采用两阶段设计:Stage I通过多智能体协作的检索增强推理快速接近可行区域,Stage II通过GRPO训练的片段级优化器进行精细调节,这种解耦设计兼顾了探索效率和精确控制能力。
核心方法
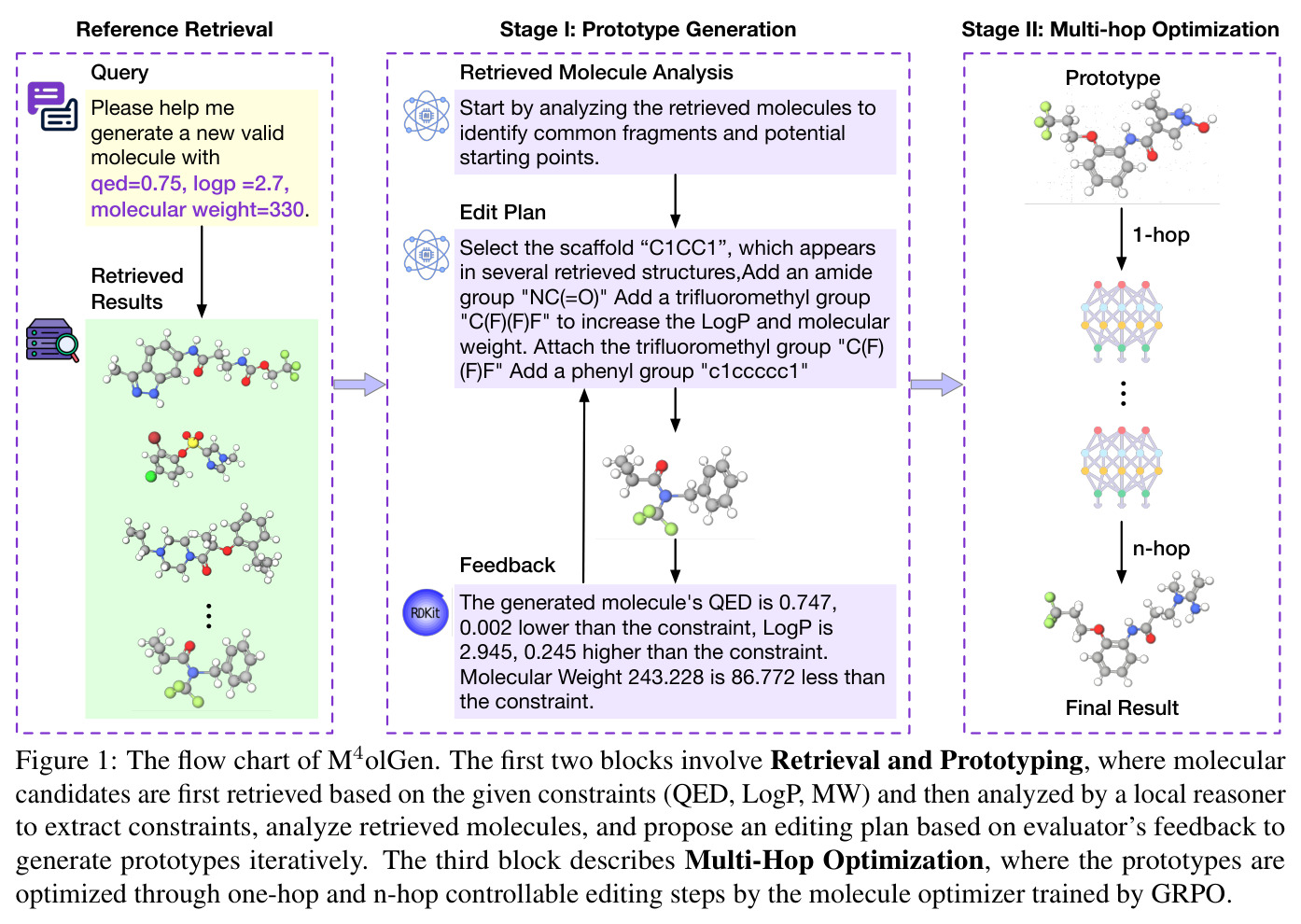
M⁴olGen是一个片段级、检索增强的两阶段框架,其核心直觉是:首先利用大规模分子数据库中的相似分子作为锚点,通过多智能体协作快速生成一个接近可行区域的原型分子;然后使用强化学习训练的优化器对原型进行精细的片段级编辑,逐步最小化与目标属性的距离。技术路线如下:Stage I执行检索增强原型生成,给定自然语言查询(如生成QED=0.75, LogP=2.7, MW=310的分子),系统首先解析出目标属性向量p_tgt,然后从大规模标注分子语料库中检索属性接近目标的参考分子集合,最后由多智能体推理器基于参考分子和RDKit反馈进行迭代的片段级编辑。Stage II应用GRPO训练的片段级优化器,以Stage I的输出为种子,执行1到3跳的精细编辑,显式最小化距离目标函数E(m) = sum_i w_i |p_i(m) - p_i^tgt|,同时通过hop预算和正则化器控制编辑复杂度和与原型的偏差。
M⁴olGen的核心创新在于将分子生成问题转化为片段编辑空间上的可验证优化问题,这一视角与已有方法有本质区别。已有LLM方法试图让模型直接理解并满足数值约束,但由于LLM缺乏精确数值推理能力,效果有限。本文的洞察是:利用RDKit等化学预言机提供快速、精确的属性反馈作为奖励信号,将强化学习的优势与片段级化学编辑的可解释性相结合。GRPO的组内相对排名机制特别适合这一场景,因为它无需ground-truth编辑序列,只需比较同一分子的不同编辑版本的奖励值即可更新策略。此外,通过BRICS片段分解将分子编辑空间离散化为可操作的添加、删除、替换操作,既保证了编辑的化学合理性(通过RDKit有效性检查),又使搜索空间保持可控。这种设计使得优化器能够在化学上有意义的子结构层面进行推理,而非在原子或SMILES字符层面盲目搜索。
方法步骤详情
M⁴olGen的方法分为以下关键步骤。Stage I:检索增强原型生成。(1)查询解析:给定自然语言查询q,规则解析器提取各属性的数值目标,形成目标属性向量p_tgt = (p_QED, p_LogP, p_MW),其中p_QED属于[0,1],p_LogP为实数,p_MW大于0。(2)参考检索:从标注分子语料库Omega中检索满足每属性容差|p_i(m) - p_i^tgt|小于等于epsilon_i的参考分子集合M,其中QED容差为正负0.05,LogP为正负0.5,MW为正负25 Da。(3)原型推理:多智能体推理器在每步迭代中选择一个动作a_t属于{replace, add, remove},基于参考分子、经验池和RDKit属性反馈进行片段编辑,生成中间分子m_t = Edit(m_{t-1}; a_t),直到距离低于阈值tau或达到最大步数T_max。(4)输出原型m_local。Stage II:GRPO片段优化。(1)片段分解:使用BRICS将原型分解为片段集合Phi(m_0) = {f_1, ..., f_k}。(2)优化器训练:以ChemDFM-v1.5-8B为基础模型,先进行5000步SFT冷启动,再进行37500步GRPO训练。奖励函数R(m) = r_format(m) + r_prop(m) - r_repeat(m) - r_invalid(m),其中属性奖励r_prop(m) = 1 - E(m),使用归一化因子alpha_q=1, alpha_l=6, alpha_w=100。(3)多跳优化:以hop预算H控制迭代次数,每跳采样8个候选(温度T=1.0, top-p=0.9, top-k=50),选择最优编辑,返回最终分子m* = m_H。
技术新颖性
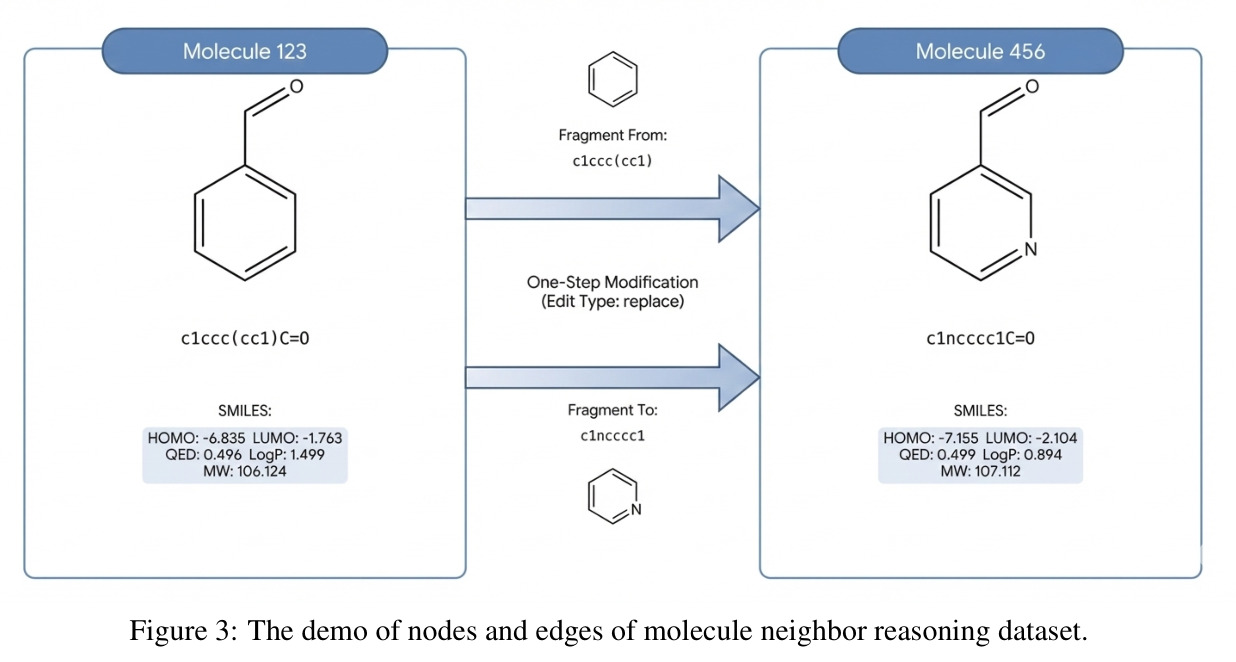
M⁴olGen的技术新颖性体现在多个维度。首先,这是首次将GRPO应用于分子生成领域的数值条件优化场景,将原本用于LLM偏好学习的组相对策略优化技术创造性地迁移到离散化学动作空间,实现了无需ground-truth演示的奖励驱动精炼。其次,本文提出了片段级多跳优化机制,通过BRICS片段分解将分子编辑操作限制在化学上有意义的子结构层面,每跳执行单个片段编辑,通过累积小的局部改进逐步逼近目标,这种设计既保证了编辑的可解释性,又通过hop预算实现了可控的复杂度调节。第三,本文构建了迄今最大的分子推理数据集(约295万分子带BRICS片段标注,约117万单编辑对),每个分子都配有显式的单跳邻居列表,记录了编辑类型、编辑片段和属性变化量(delta_QED, delta_LogP, delta_MW),为可控推理链的构建提供了数据基础。第四,多智能体协作的检索增强原型生成设计将领域知识(通过参考分子)、经验(通过邻居对/树)和工具反馈(通过RDKit)三源信息整合,实现了高效的可行区域定位。
实验结果
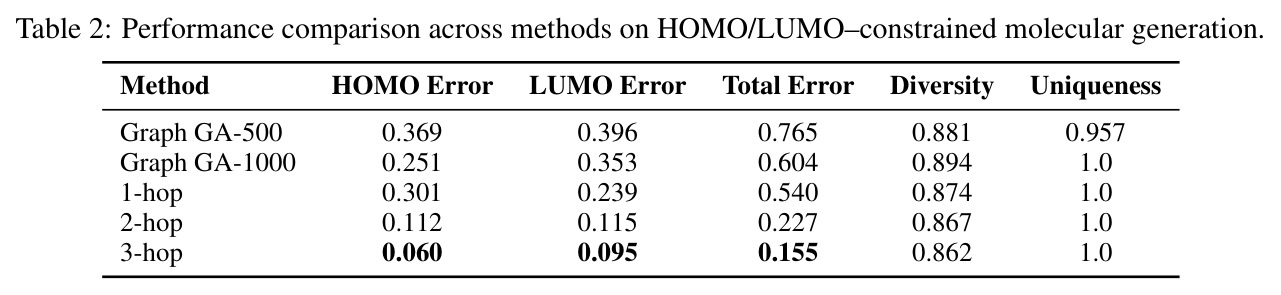
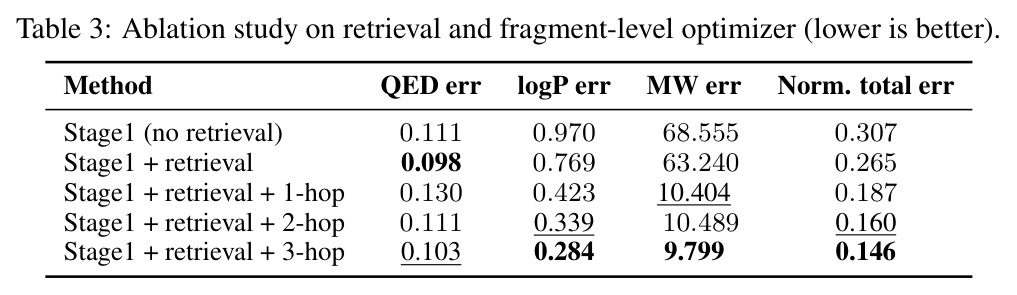
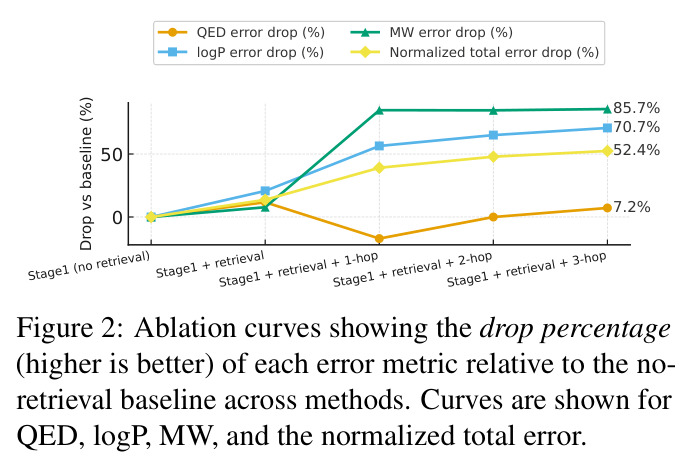
M⁴olGen在两个属性集上进行了全面实验,取得了显著的性能提升。QED/LogP/MW约束实验:在100个均匀采样的目标元组上,每个基线运行10次独立实验取最优,3-hop-GPT4o配置达到最低归一化总误差(NTE)0.146,相比最强商业模型GPT-4.1(NTE 0.255)提升42.7%。具体来看,QED误差为0.103,LogP误差为0.284(相比STGG-50x的0.566降低49.8%),MW误差为9.799(相比STGG-50x的63.917降低84.7%)。多样性和唯一性指标分别为0.884和1.0,与最强图基线Graph GA-1000(0.886, 1.0)相当。HOMO/LUMO约束实验:在HOMO和LUMO能量属性约束下,M⁴olGen同样表现优异。3-hop配置实现总误差0.155,其中HOMO误差0.060,LUMO误差0.095,两者同时降低表明实现了平衡的多属性优化。相比之下,Graph GA-1000的总误差为0.604,M⁴olGen实现了近4倍的改进,且推理时间减少约90%。消融实验:消融研究验证了各组件的贡献。添加检索使归一化总误差从0.307降至0.265(降低13.7%),主要由LogP(20.7%降低)和MW(7.8%降低)的改进驱动。引入片段优化器产生最大改进,从检索基线的0.265降至1-hop的0.187(39.1%降低),进一步降至3-hop的0.146(52.4%降低)。NTE随hop数单调递减,验证了多跳机制的有效性。值得注意的是,Qwen3-14B-chem配置也取得竞争性结果(NTE 0.159),表明方法不依赖商业模型。
查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| QED/LogP/MW三属性约束分子生成 | 归一化总误差(NTE) | 0.146(3-hop-GPT4o) | GPT-4.1: 0.255, STGG+: 0.155, Graph GA-1000: 0.187 | 相比GPT-4.1提升42.7%,相比STGG+提升5.8% |
| HOMO/LUMO能量属性约束生成 | 总误差(eV) | 0.155(3-hop) | Graph GA-500: 0.765, Graph GA-1000: 0.604 | 相比Graph GA-1000提升74.3% |
| QED单属性精度 | QED MAE | 0.103(3-hop-GPT4o) | STGG+: 0.079, GPT-4.1: 0.115 | 竞争性结果,优于GPT-4.1 10.4% |
| LogP单属性精度 | LogP MAE | 0.284(3-hop-GPT4o) | STGG+: 0.418, GPT-4.1: 0.697 | 相比STGG+提升32.1% |
| MW单属性精度 | MW MAE(Da) | 9.799(3-hop-GPT4o) | Graph GA-1000: 7.95, STGG+: 23.56 | 相比STGG+提升58.4% |
| 生成多样性 | ECFP4 Tanimoto多样性 | 0.884 | Graph GA-1000: 0.886, STGG+: 0.879 | 与最强基线相当 |
局限与改进
本文存在几个值得讨论的局限性。首先,作者坦诚指出研究依赖于计算属性(如RDKit估算器)而非实验测量值,这意味着生成的分子在实际合成中可能表现出与预测不同的属性。其次,评估的属性集相对有限,仅涵盖QED、LogP、MW、HOMO、LUMO五个属性,而实际药物发现涉及更多复杂属性(如ADMET性质、毒性、合成可及性等)。第三,虽然更深层的hop持续改进性能,但计算成本显著增加且收益递减,这是一个实际的设计权衡——从1-hop到2-hop NTE从0.187降至0.160(降低14.4%),但从2-hop到3-hop仅从0.160降至0.146(降低8.8%)。第四,QED在1-hop阶段出现小幅回退(从0.098升至0.130),表明多目标优化中存在属性间的权衡,虽然在3-hop时恢复到0.103,但这反映了精确多属性控制的内在难度。第五,数据集构建依赖于BRICS片段分解的启发式规则,可能无法覆盖所有化学上合理的编辑操作。最后,Stage I使用GPT-4o作为推理LLM,引入了商业API依赖和成本问题。
独立分析的弱点
M⁴olGen存在几个值得深入分析的弱点。属性集局限性:当前仅验证了5个属性(QED、LogP、MW、HOMO、LUMO),而实际药物发现涉及更复杂的ADMET性质(吸收、分布、代谢、排泄、毒性)和合成可及性。改进方向是扩展到更多属性类型,特别是需要实验验证的属性。计算效率:多跳优化的推理成本随hop数线性增长,且Stage I的LLM推理本身就是主要瓶颈。可以探索更高效的优化策略,如自适应hop数选择或早停机制。片段编辑空间受限:BRICS分解基于启发式规则,可能遗漏化学上合理但不在BRICS规则内的编辑。可以引入更灵活的片段化方法或学习化的编辑空间。Stage I与Stage II的解耦:两个阶段分别优化,可能存在信息损失。可以探索端到端训练或更紧密的梯度连接。评估协议:best-of-10的评估方式可能高估了单次生成的成功率,实际部署时需要考虑单次生成的可靠性。
未来方向
基于本文成果,未来研究可沿以下方向展开。更丰富的属性扩展:将框架扩展到蛋白质-分子对接、自由能微扰等需要昂贵计算的属性,验证GRPO在稀疏奖励场景下的效果。自适应hop策略:开发动态hop数选择机制,根据当前分子与目标的距离自动决定是否需要更多编辑,避免不必要的计算。更精细的编辑操作:超越BRICS片段,引入原子级别的精细编辑,或结合逆合成分析确保生成分子的合成可及性。多模态融合:结合3D分子构象信息,将框架扩展到构象敏感的属性优化。实际验证:与药物化学家合作,将生成的分子进行实际合成和生物活性测试,验证计算属性与实验值的一致性。在线学习:开发在线RL机制,允许优化器在部署过程中根据实际反馈持续改进。作者提出的扩展:论文提到可以向更丰富的预言机扩展,随着更真实发现设置的扩展,方法可处理更复杂的属性和更大的搜索空间。
复现评估
本文的复现性评估如下。数据开源:论文声称构建了大规模公开数据集(约295万分子带BRICS片段标注,约117万单编辑对),但未明确说明数据集是否已公开发布及获取方式。数据来源于ZINC、CHEMBL和MOSES三个公开数据库的组合。模型与代码:Stage II使用ChemDFM-v1.5-8B作为基础模型,该模型可在HuggingFace获取。但论文未提及代码是否开源,这可能影响完全复现。算力需求:Stage II的GRPO训练在单块NVIDIA A100(40GB)上进行,SFT 5000步加GRPO 37500步,这是中等规模的计算需求,学术实验室通常可以负担。Stage I使用GPT-4o API,需要相应的API访问权限和费用。关键超参数:论文提供了详细的训练参数(采样8个候选,温度1.0,top-p=0.9,top-k=50,归一化因子alpha_q=1, alpha_l=6, alpha_w=100),有利于复现。评估协议:100个目标元组乘以10次实验的评估协议清晰明确,但需要能够访问相同的分子数据库来构建检索索引。总体而言,中等复现难度,主要障碍可能是代码开源情况和Stage I的API依赖。
论文图表