AceFF:面向小分子的前沿机器学习原子间势能模型 AceFF: A State-of-the-Art Machine Learning Potential for Small Molecules
TensorNet2架构的药物小分子机器学习力场,兼顾速度与DFT级精度。
前置知识
机器学习原子间势能(MLIP)
MLIP是用神经网络拟合量子力学计算数据来预测原子间相互作用的方法。它以原子坐标和原子序数为输入,通过消息传递图神经网络学习原子间的能量和力。典型流程是:先用DFT等量子力学方法计算大量分子的构型-能量-力数据,再训练神经网络模型。推理时,模型能以接近DFT的精度计算能量和力,但速度提升数个数量级——比DFT快$10^3$-$10^6$倍,只比经典分子力场慢约一个数量级。
本文的核心就是提出一个新的MLIP模型,理解MLIP的范式是阅读本文的基础。
密度泛函理论(DFT)
DFT是一种量子力学计算方法,通过电子密度来描述多电子体系的基态性质。在分子模拟中,DFT能提供较高精度的能量和力的计算,但计算成本随体系大小呈$O(N^3)$到$O(N^4)$增长。文中使用的DFT级别为$\omega$B97M-V/def2-TZVPPD,这是一种高精度的泛函和基组组合,能给出可靠的分子能量参考值。
DFT是本文训练数据的生成方法,也是评估模型精度的量子力学基准。理解DFT精度与计算代价之间的权衡是理解MLIP存在意义的关键。
TensorNet架构
TensorNet是一种等变消息传递图神经网络架构。它为每个原子学习一个$3\times3$矩阵(二阶张量)$\mathbf{X}^{(i)}$,该表示在$O(3)$旋转和翻译下具有等变性。张量可分解为标量$I$、矢量$\mathbf{A}$和对称无迹张量$\mathbf{S}$三个分量,分别对应1、3、5个自由度。消息传递过程中,每个原子的张量表示根据邻居原子的信息进行更新。最终通过计算张量的Frobenius范数的平方得到旋转不变的特征,再经MLP预测原子能量,力则通过自动微分获得。
TensorNet是AceFF的基础架构,TensorNet2是在其上的改进。理解TensorNet的张量表示和消息传递机制是理解本文方法创新的前提。
电荷平衡与库仑相互作用
在分子体系中,原子带有部分电荷(partial charges),这些电荷之间的库仑相互作用对分子能量有重要贡献。传统MLIP只在短程截断范围内工作(通常5-6 Angstrom),无法捕捉长程静电效应。电荷平衡(charge equilibration)是一种让预测的部分电荷满足总电荷约束的方法:对于总电荷为$Q$的分子,所有原子的部分电荷之和必须等于$Q$。AIMNet2提出了一种中性电荷平衡(Neutral Charge Equilibration, NQE)方案,通过在每个特征通道独立进行电荷平衡来实现这一点。
TensorNet2的核心创新就是将AIMNet2的电荷平衡机制引入TensorNet,以正确处理带电分子和长程静电相互作用。
CUDA图与推理优化
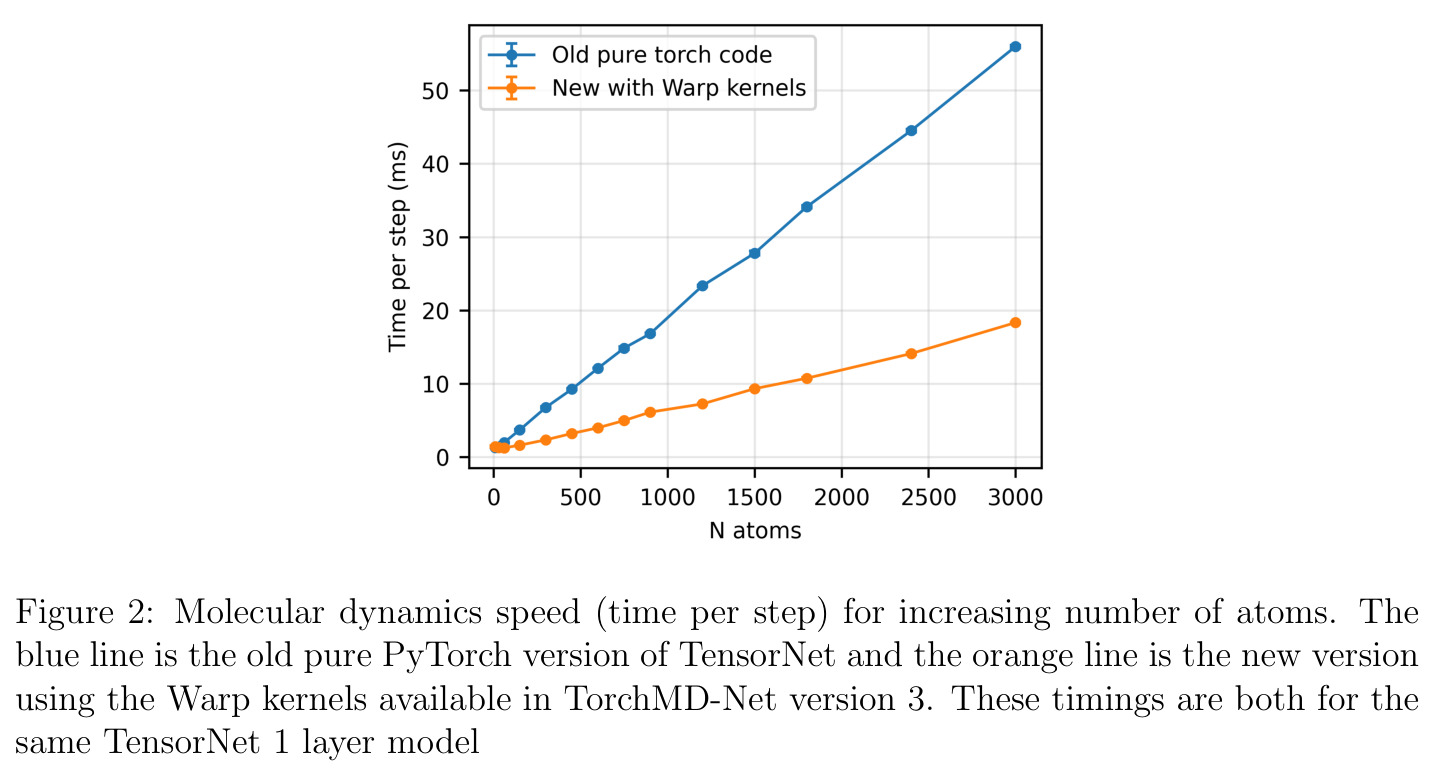
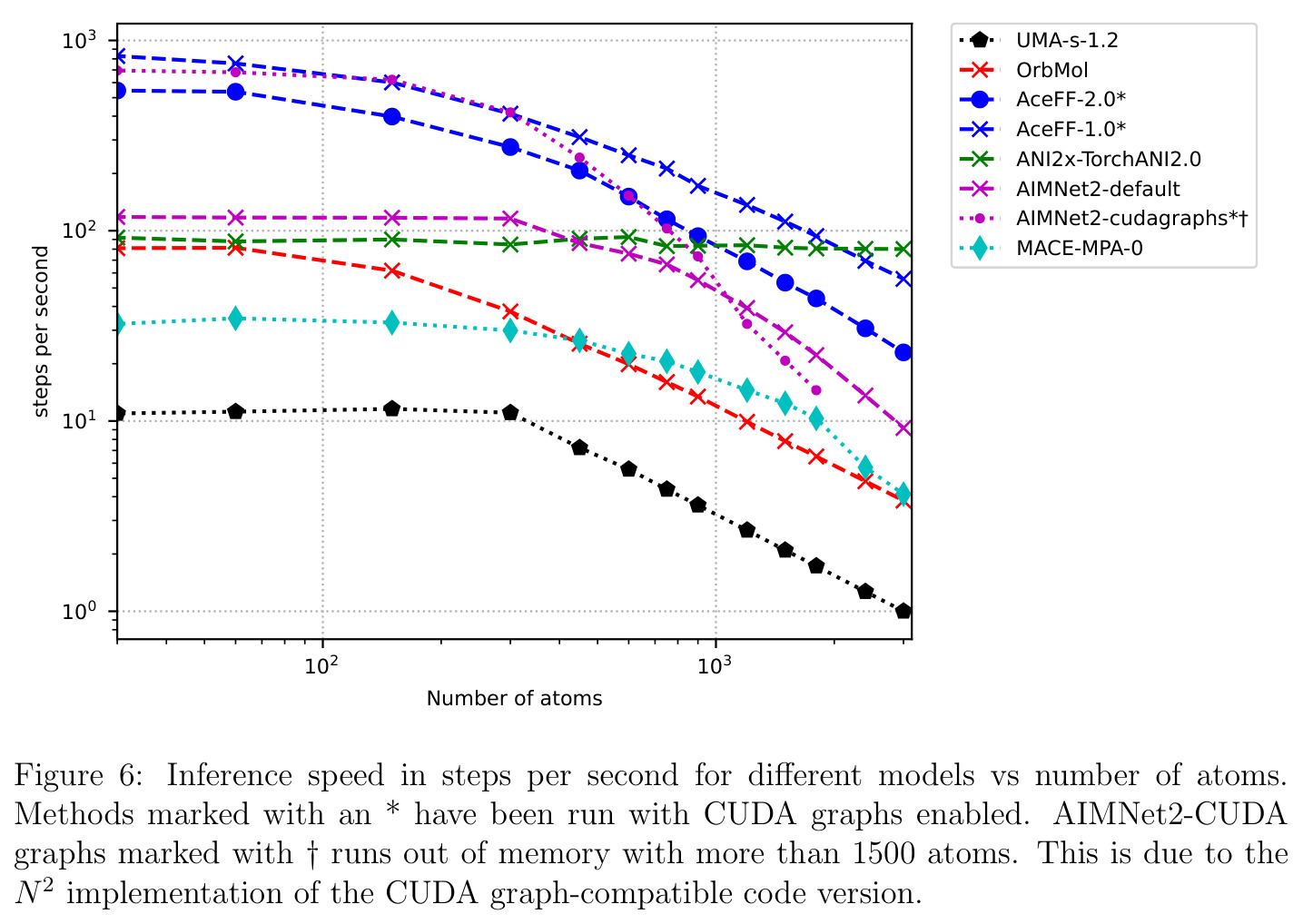
CUDA图(CUDA Graphs)是NVIDIA提供的一种GPU执行优化技术,允许将一系列GPU操作记录为一个图,然后一次性提交执行,避免了CPU-GPU之间的频繁同步开销。对于小体系(<300原子)的分子动力学模拟,GPU kernel的启动延迟是主要瓶颈,CUDA图可以将延迟从10ms以上降低到1ms以下。这使得小体系能达到100+步/秒的模拟速度。此外,文中还使用了NVIDIA Warp自定义内核来加速张量分解/组合和消息传递操作,实现了3倍的速度提升和3倍的GPU内存节省。
软件优化是本文的重要贡献之一,理解CUDA图和Warp内核对推理速度的影响对于评估模型的实际应用价值至关重要。
扭转扫描(Torsion Scan)
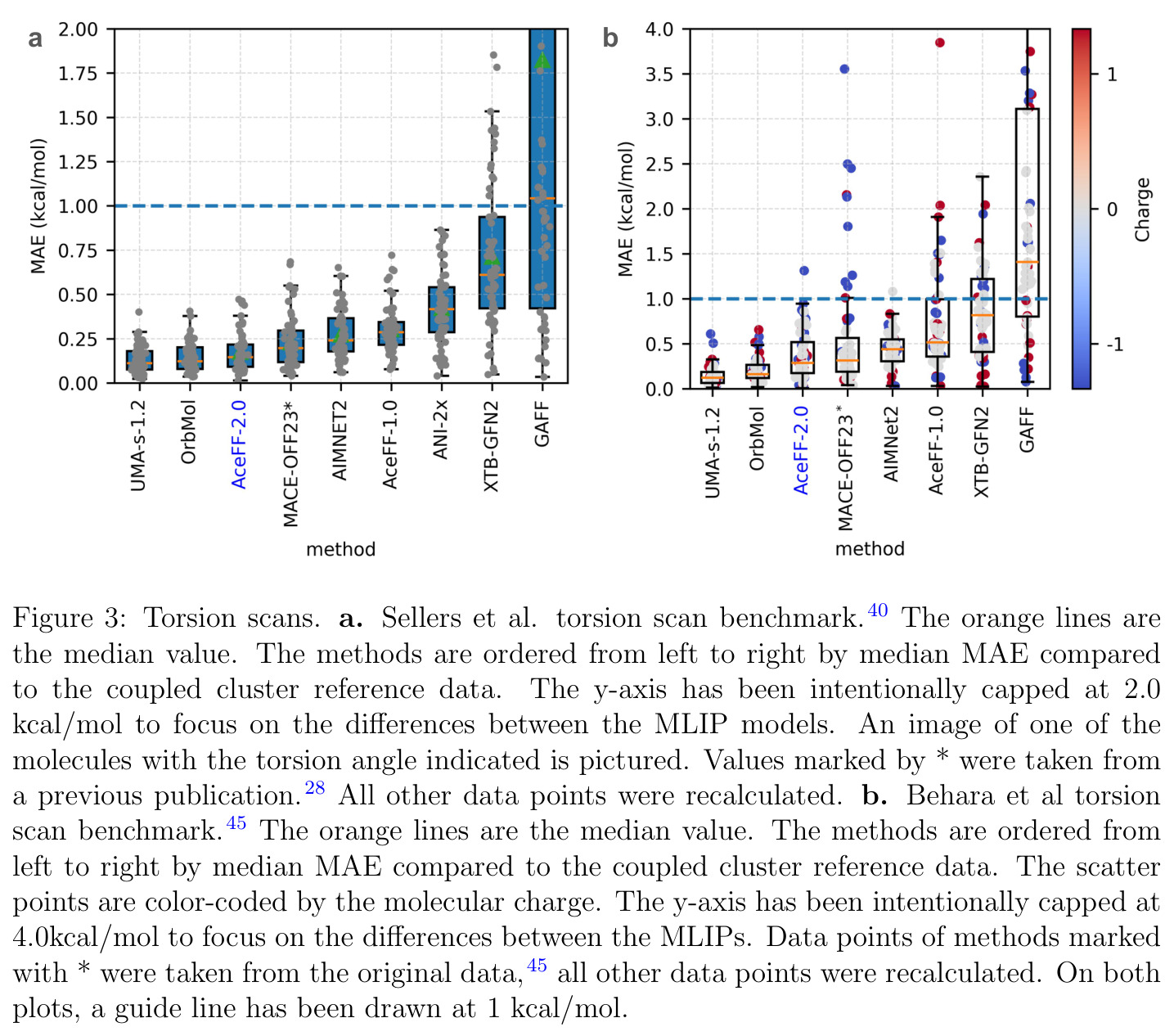
扭转扫描是评估力场精度的常用方法。它固定分子中的一个二面角,然后以一定步长旋转该角度,在每个角度下进行几何优化(或单点计算),得到势能随扭转角度变化的曲线。这条曲线对于药物发现至关重要,因为分子的构象柔性直接影响其与蛋白质的结合亲和力。评估时以CCSD(T)/CBS等高精度量子化学方法的结果为参考,计算各方法预测曲线的平均绝对误差(MAE)。
扭转扫描是本文的核心基准测试之一,直接关系到MLIP在药物发现构象采样中的可靠性。
研究动机
药物发现中的分子模拟面临一个根本性困境:经典分子力学力场(如GAFF、CGenFF、OpenFF)虽然计算效率高,但在处理多样化的药物样分子时预测精度不足,尤其是对于含有罕见官能团、需要考虑量子效应或极化效应的分子。以GAFF为例,在Behara等人的扭转扫描基准测试中,其MAE远超所有MLIP模型,在Wiggle150基准上MAE高达22.87 kcal/mol。另一方面,第一性原理量子力学方法(如DFT)虽然精度高,但计算成本随体系大小呈$O(N^3)$增长,对于常规生物分子模拟来说代价过高。现有的MLIP模型也各有缺陷:ANI-2x仅支持8种元素且只处理中性分子,严重限制了其在药物化学中的应用——药物分子常含有卤素(F、Cl、Br、I)、硫、磷等元素,且经常带电荷;MACE-OFF23模型虽然准确但推理速度较慢;OrbMol和UMA虽然覆盖范围广但对小分子药物发现而言不够快速。
本文的目标是本文的具体目标是开发一个专门为小分子药物发现优化的MLIP模型——AceFF-2,使其同时满足以下要求:(1)支持药物化学中所有必需元素(H、B、C、N、O、F、Si、P、S、Cl、Br、I共12种),覆盖周期表中几乎所有常见有机元素;(2)能够正确处理带电分子(电荷范围从-2到+2),因为许多药物分子和中间体在生理pH下是带电的;(3)在推理速度上接近甚至超过ANI-2x,使其能够实际用于大规模的相对结合自由能(RBFE)计算等药物发现工作流;(4)在精度上达到DFT水平,特别是在扭转能扫描、力预测和分子动力学稳定性方面。
与已有工作不同的是,本文的独特切入角度体现在三个方面。首先,在架构层面,作者将AIMNet2的电荷平衡机制创造性地整合到TensorNet架构中,命名为TensorNet2。这不是简单的拼接,而是在每个消息传递层后都引入电荷预测和平衡步骤,使得电荷信息能够参与后续的消息传递更新,形成一种自洽的电荷预测机制。其次,在数据层面,作者构建了一个包含约200万独特分子、1200万构象的DFT数据集,特别强调包含了高能构型,因为他们认为这对于训练稳定的MLIP至关重要——这不同于OMol25等通用数据集的设计理念。最后,在软件优化层面,作者利用NVIDIA Warp自定义内核对TensorNet的核心操作进行了底层优化,实现了3倍速度提升和3倍内存节省,这使得TensorNet2成为最快的等变MLIP之一。
核心方法
AceFF-2的整体技术路线可以这样理解:首先,作者认识到现有的TensorNet架构虽然速度快,但在处理带电分子时存在外推困难——当测试较大的带电分子时,其性能显著落后于AIMNet2。因此,他们借鉴AIMNet2的电荷处理思想,在TensorNet的每个消息传递层中引入电荷预测和中性电荷平衡机制,形成了TensorNet2架构。然后,使用内部构建的DFT数据集($\omega$B97M-V/def2-TZVPPD级别)训练了一个2层TensorNet2模型,包含128维隐藏维度、32个径向基函数和16维电荷嵌入。同时,作者对TorchMD-Net包中的TensorNet实现进行了底层软件优化,使用NVIDIA Warp自定义内核替代了原有的PyTorch实现,获得了3倍速度提升。最后,通过六个不同的基准测试全面评估模型在扭转能精度、应变构型能量、力预测、势能面平滑度、推理速度和分子动力学稳定性等方面的表现。
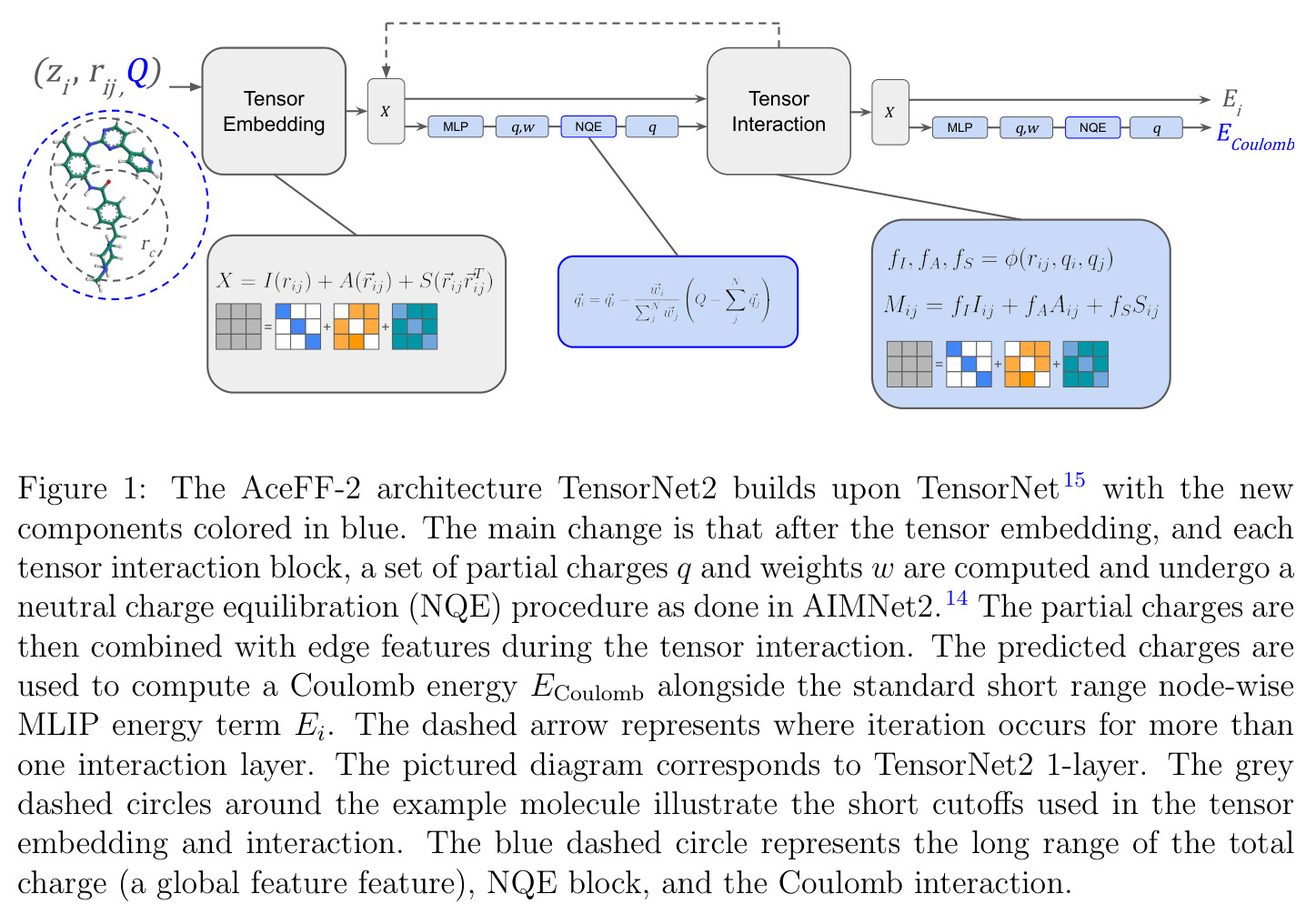
TensorNet2的核心创新是在TensorNet的等变消息传递框架中嵌入了AIMNet2风格的中性电荷平衡(NQE)机制。这一创新的本质区别在于:传统的TensorNet只将总分子电荷$Q$作为一个简单的全局特征引入,没有额外的学习参数,虽然能解决具有相同原子坐标但不同电荷的分子退化问题,但在外推到更大的带电分子时表现不佳。而TensorNet2在每个消息传递层后都引入一个电荷预测神经网络,从节点特征$(I_i, \mathbf{A}_i, \mathbf{S}_i)$输出一组电荷预测$\vec{q}_i$和权重$\vec{w}_i$。关键公式为:$$\vec{q}_i = \vec{q}_i - \frac{\vec{w}_i}{\sum_j \vec{w}_j} \left(Q - \sum_j \vec{q}_j\right)$$这个等式独立地在每个特征通道上执行电荷平衡,确保部分电荷之和等于总分子电荷$Q$。更重要的是,这些预测的电荷被用于扩展标量边特征,使得消息传递公式变为:$$f^I_i, f^A_i, f^S_i = \phi(r_{ij}) \cdot \text{SiLU}(\text{MLP}(e_{\text{RBF}}(r_{ij}) \oplus q_i \oplus q_j))$$其中$\oplus$表示张量拼接。这意味着电荷信息不仅在最终层使用,而是在每一层都参与消息传递,形成了一种自洽的电荷预测过程。各层的电荷预测还被用于计算加权库仑能$E_{\text{Coulomb}}$,权重设定使得最后一层的输出对总能量影响最大。这种方法相比4G-HDNNP等需要求解线性方程组的方案,计算开销很小——参数仅增加28%,速度仅下降25%。
方法步骤详情
AceFF-2的完整方法可以分为以下几个步骤:(1)数据准备:从PubChem中选取约200万独特药物样分子,包含H、B、C、N、O、F、Si、P、S、Cl、Br、I共12种元素,分子大小最多30个原子,电荷范围[-2, 2]。使用PySCF和GPU4PySCF在$\omega$B97M-V/def2-TZVPPD级别进行DFT计算,生成约1200万个构象的能量和力标签,数据集包含能量最小化结构和高能几何构型。(2)架构设计:在TensorNet基础上构建TensorNet2。输入原子序数$Z_i$和坐标$\mathbf{R}_i$,首先进行张量嵌入,为每个原子学习一个等变的$3\times3$矩阵$\mathbf{X}^{(i)}$。在每个消息传递层:a) 将张量分解为$I, \mathbf{A}, \mathbf{S}$三个分量(分别1、3、5个自由度,而非原始的3个$3\times3$矩阵);b) 电荷预测网络从节点特征输出电荷预测$\vec{q}_i$和权重$\vec{w}_i$;c) 执行中性电荷平衡确保$\sum_i q_i = Q$;d) 将电荷信息扩展到边特征中进行消息传递更新。(3)能量计算:每个原子的短程能量$E_i$通过张量表示的Frobenius范数平方经MLP预测;同时从各层的电荷预测计算加权库仑能$E_{\text{Coulomb}}$作为长程能量项。总能量$E = \sum_i E_i + E_{\text{Coulomb}}$。力通过自动微分$\mathbf{F}_i = -\partial E / \partial \mathbf{R}_i$获得。(4)训练:使用TorchMD-Net的torchmd-train脚本,2层TensorNet2,128维隐藏维度,32个RBF,16维电荷嵌入,共685,413个参数。使用能量和力标签训练,不使用部分电荷或偶极矩数据。(5)软件优化:使用NVIDIA Warp自定义内核优化TorchMD-Net中的核心操作,包括$I, \mathbf{A}, \mathbf{S}$张量的分解/组合以及消息传递例程,实现紧凑格式(1+3+5=9个值而非3个$3\times3$=27个值)。
技术新颖性
TensorNet2的技术新颖性体现在多个层面。首先,在架构创新上,它是首个将AIMNet2风格的自洽电荷平衡机制与等变张量网络结合的模型。不同于Latent Ewald Summation方法只在最终节点特征上计算库仑相互作用,TensorNet2在每一层都进行电荷预测和平衡,使得电荷信息能够深度参与消息传递过程。也不像4G-HDNNP方法需要求解线性方程组(计算量随体系大小增长),TensorNet2的NQE只需简单的减法和加权平均操作,几乎不增加计算开销。其次,在等变性分析上,作者指出TensorNet的交互层自然地从$O(3)$等变性过渡到$SO(3)$等变性——张量-张量交互积混合了偶数和奇数宇称,打破了反射等变性但保持旋转等变性。这在药物发现中特别有价值,因为手性分子(对映体通过反射相关)在结合亲和力和生物活性上可能有截然不同的表现。第三,在软件实现上,作者认识到$I, \mathbf{A}, \mathbf{S}$可以分别用1、3、5个值紧凑表示,而非原来的3个$3\times3$矩阵,通过Warp内核实现了3倍速度提升和3倍内存节省,同时保持CUDA图兼容性——这是实现小体系低延迟模拟的关键。
实验结果
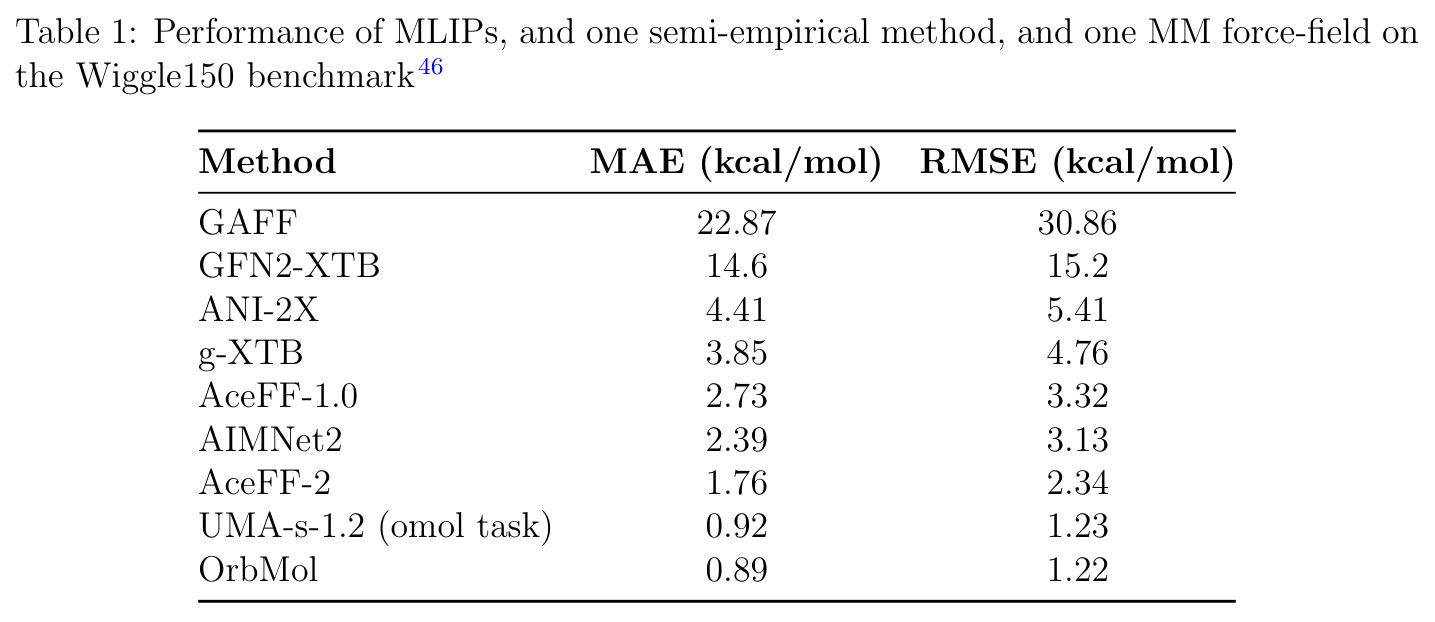
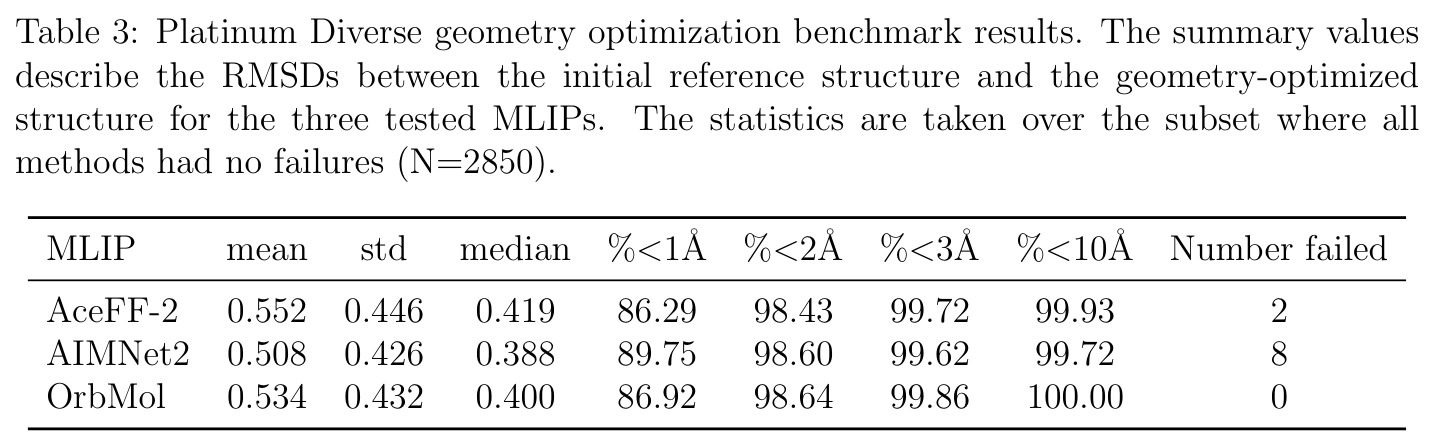
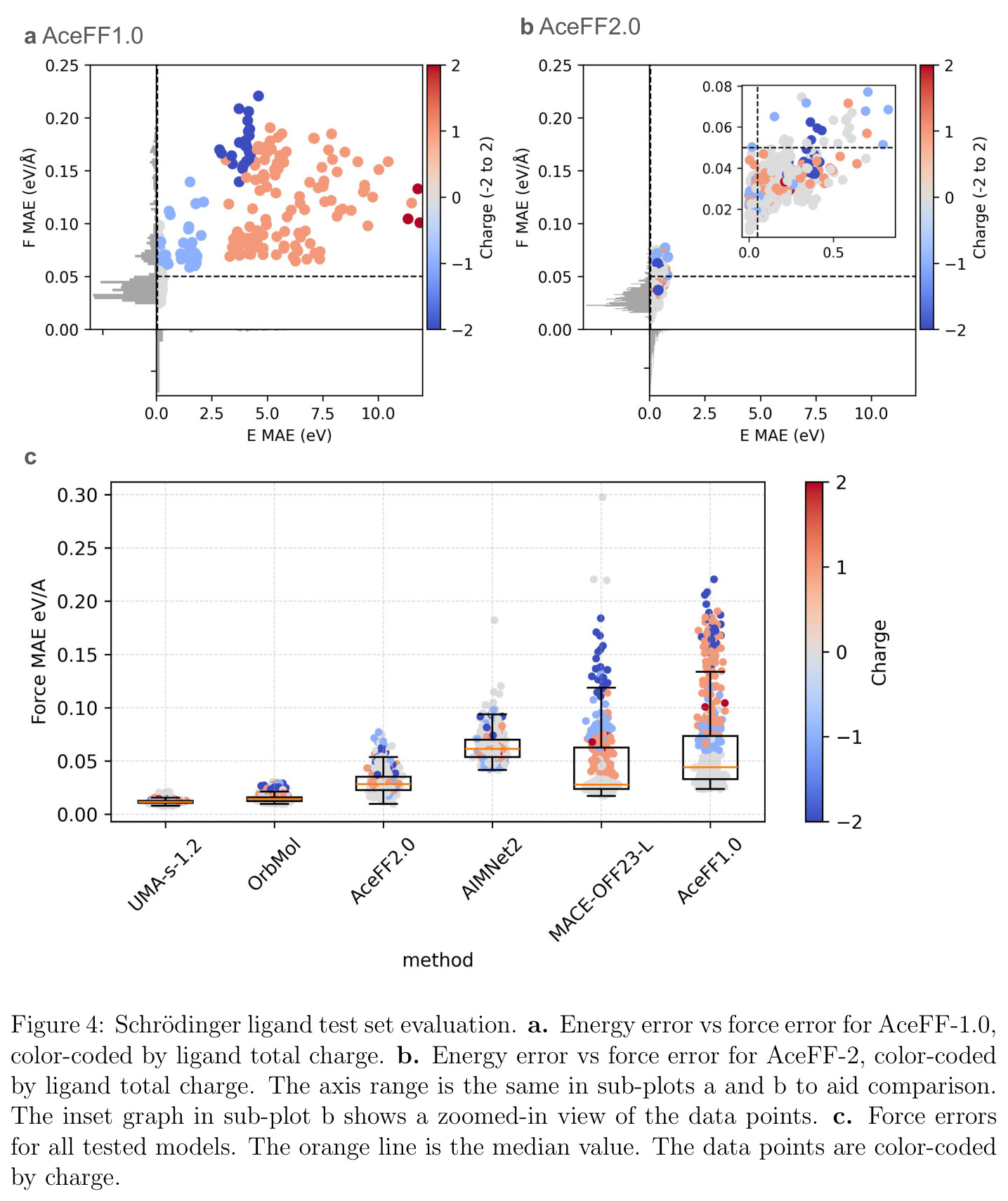
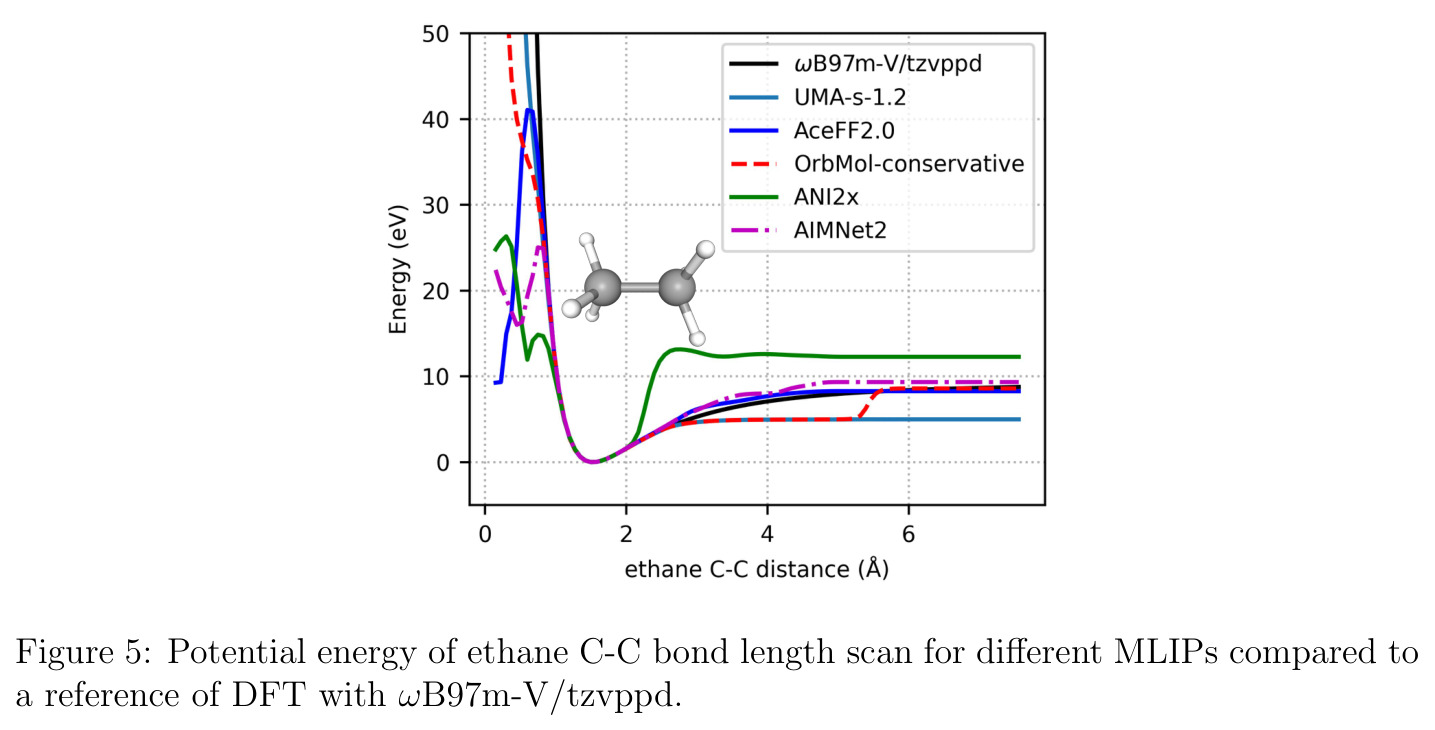
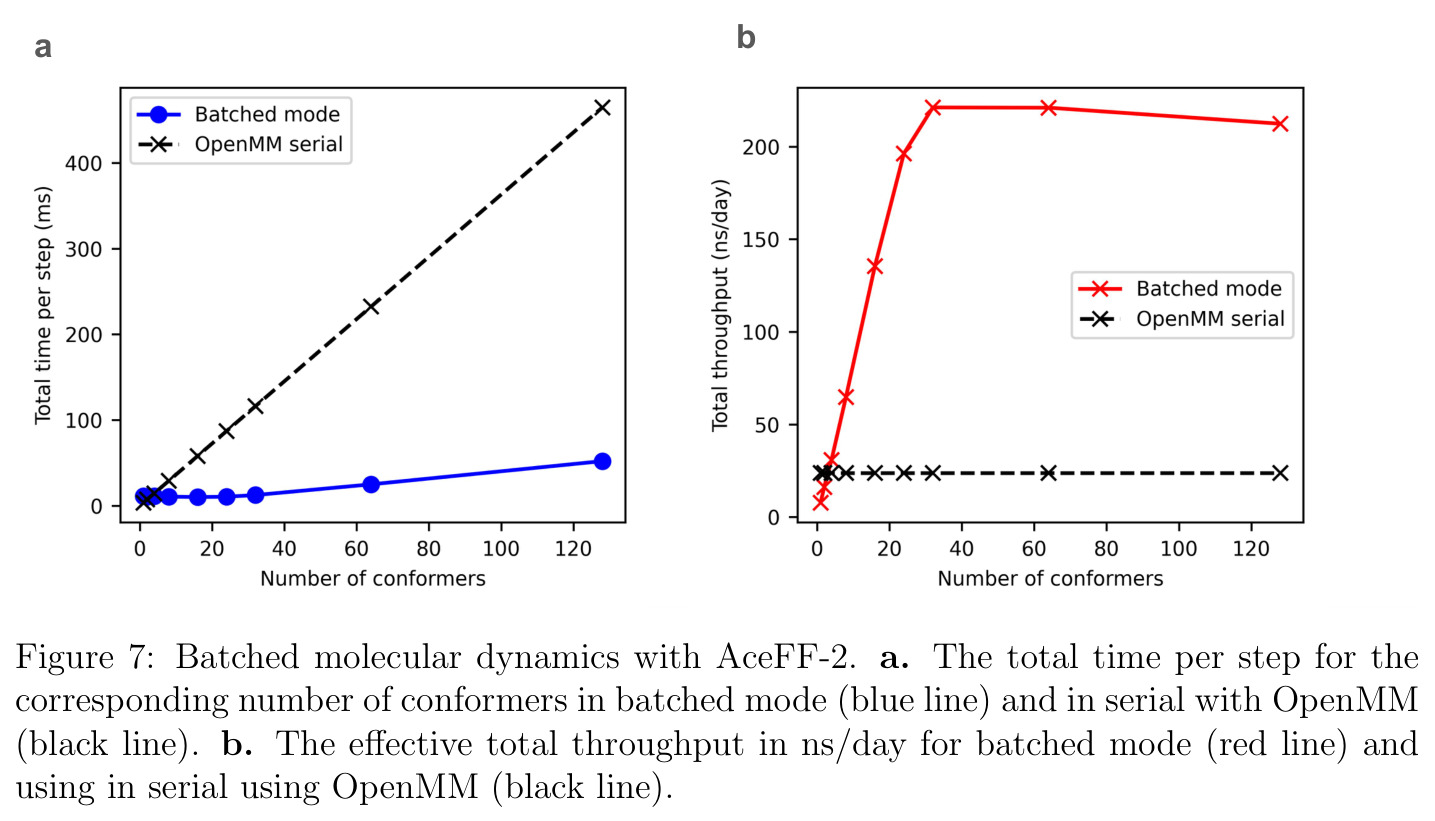
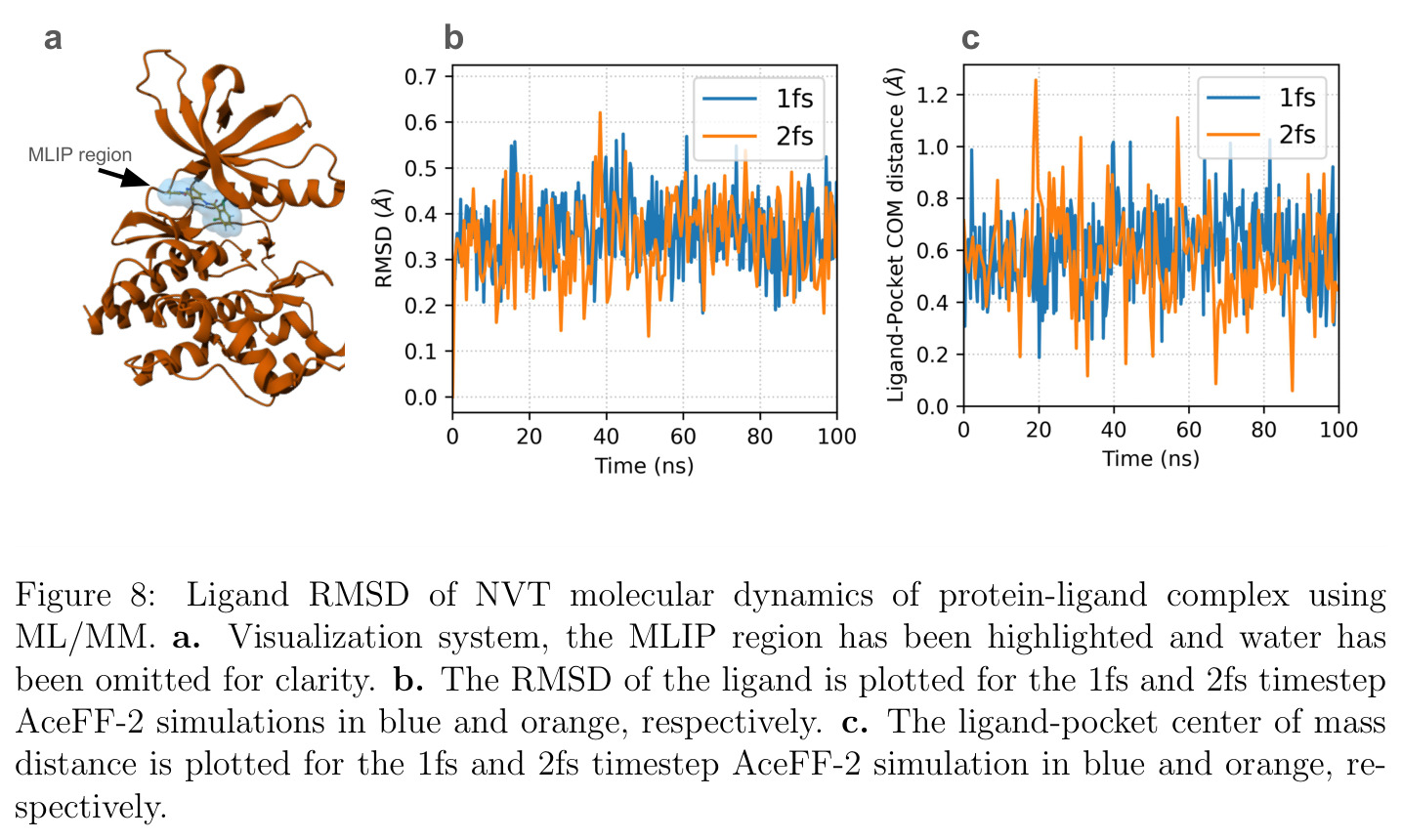
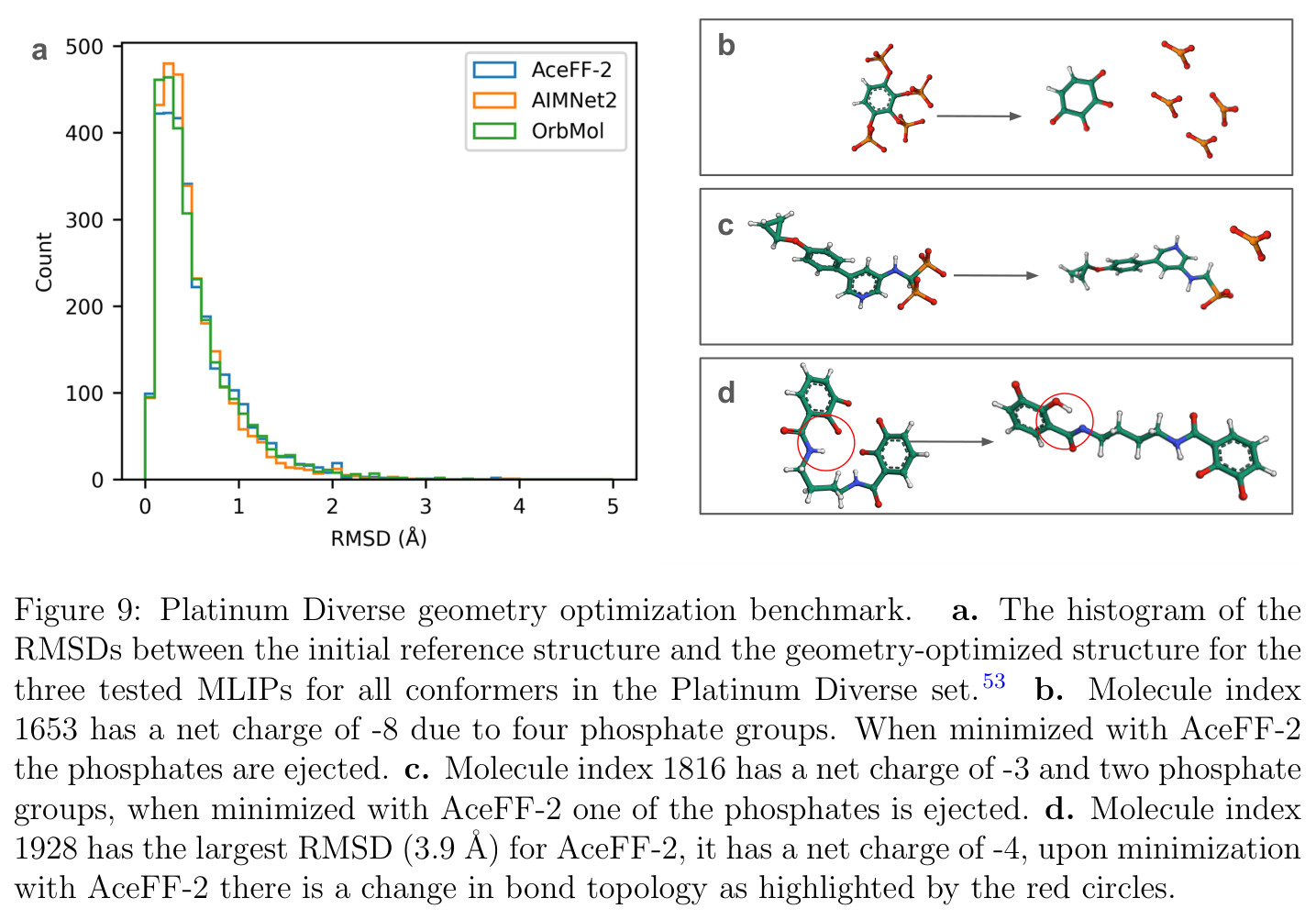
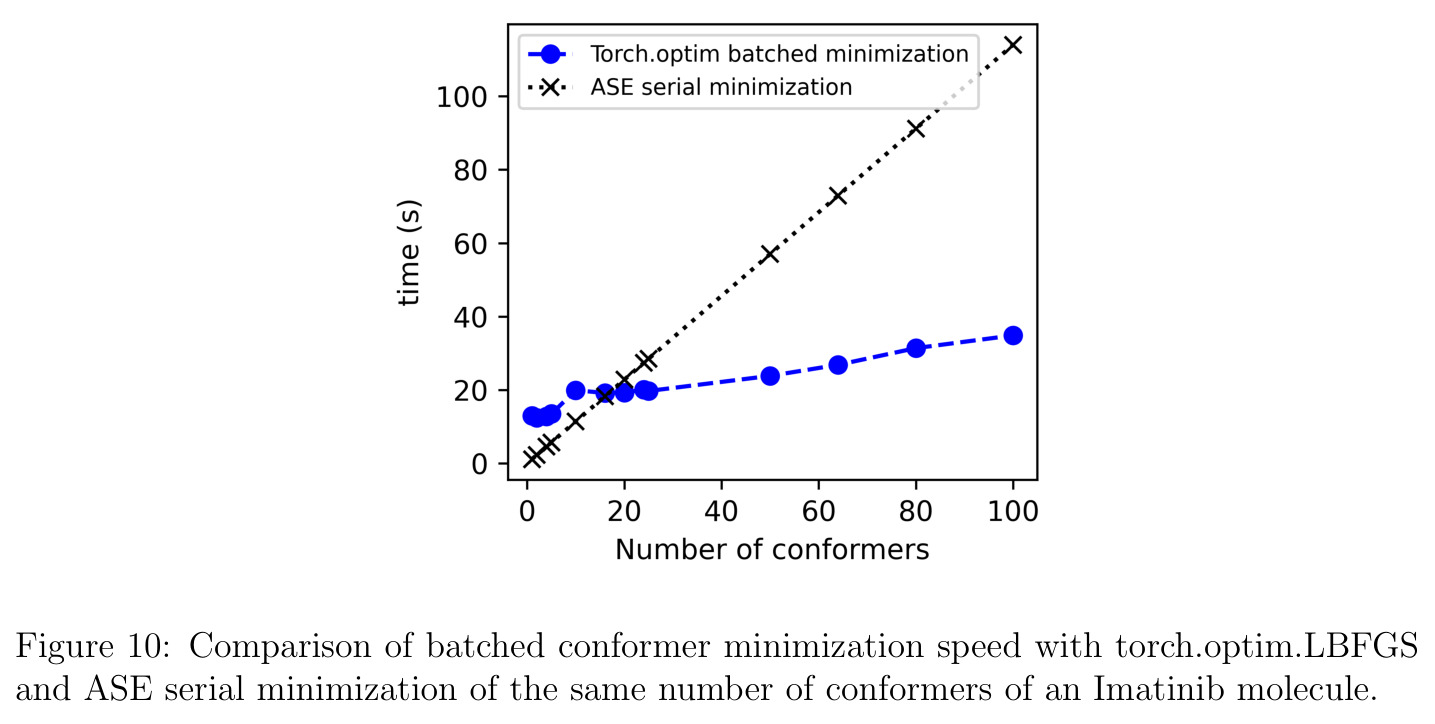
AceFF-2在六个基准测试中展现了全面且优异的表现。在扭转扫描基准测试中(Sellers等人,62个分子,参考为CCSD(T)/CBS),OrbMol的中位MAE最低,AceFF-2紧随其后,两者差距很小;而ANI-2x显著落后,GFN2-XTB表现较差,GAFF误差巨大。在带电分子的扭转扫描中(Behara等人,包含中性、-1和+1电荷分子,参考为MP2/heavy-aug-cc-pVTZ//CCSD(T)/CBS),趋势类似,但AceFF-1.0在带电分子上表现很差,MACE-OFF23也因只支持中性分子而有较大偏差;AceFF-2、UMA和OrbMol对中性和带电分子都表现更均衡。在Wiggle150基准(150个高度应变的构型,参考为DLPNO-CCSD(T)/CBS)上,OrbMol的MAE最低为0.89 kcal/mol,UMA为0.92,AceFF-2为1.76(相比AceFF-1.0的2.73有显著提升),AIMNet2为2.39,所有MLIP(除ANI-2x外)都优于半经验方法g-XTB(3.85)。在Schrodinger配体测试集(650个构象,>30个原子的药物样分子)上,AceFF-2的力误差大部分低于0.05 eV/A,远优于AceFF-1.0在带电分子上的表现;UMA表现最佳(所有力误差<0.02 eV/A),OrbMol次之(<0.03 eV/A),但需注意两者可能在训练集中见过类似分子。在乙烷C-C键扫描的势能平滑度测试中,AceFF-2匹配DFT能量至高达40 eV,仅OrbMol(含ZBL排斥项)能显示核排斥特征,而其他模型完全由神经网络建模。在分子动力学速度测试中,对于<300原子的小体系,只有AceFF和AIMNet2-CUDA图能达到>100步/秒的速度,这得益于CUDA图的低延迟特性;对于3000原子的水盒子,各模型按预期缩放。在MLIP/MM混合动力学模拟中(100 ns,RTX4090),AceFF-2以2fs时间步长达到36.7 ns/day,1fs时间步长达到72.6 ns/day,轨迹稳定。在批量构象最小化基准(Platinum Diverse数据集,2859个晶体结构)上,AceFF-2平均RMSD为0.552 A,86.29%在1 A以内,仅2个分子(电荷-8和-3的磷酸盐分子)最小化失败;AIMNet2有8个失败但平均RMSD略低(0.508 A);OrbMol无失败但平均RMSD为0.534 A。批量最小化方面,对于20个以上构象,批量模式比串行快得多,32个构象时吞吐量提升近10倍。

查看结构化数据
| 任务 | 指标 | 本文 | 基线 | 提升 |
|---|---|---|---|---|
| 扭转扫描(Sellers基准,62分子) | 中位MAE (kcal/mol) | AceFF-2紧随OrbMol(第二低) | OrbMol最低,ANI-2x显著较高 | 相比AceFF-1.0明显提升 |
| 扭转扫描(Behara基准,含带电分子) | 中位MAE (kcal/mol) | AceFF-2对中性和带电分子均表现良好 | AceFF-1.0在带电分子上偏差大,MACE-OFF23亦然 | 解决了带电分子外推问题 |
| Wiggle150应变构型(DLPNO-CCSD(T)/CBS参考) | MAE (kcal/mol) | 1.76 | OrbMol 0.89, UMA 0.92, AceFF-1.0 2.73, AIMNet2 2.39, ANI-2x 4.41, g-XTB 3.85 | 相比AceFF-1.0提升35%(2.73到1.76) |
| Schrodinger配体力预测(650构象,>30原子) | 力MAE (eV/A) | 大部分<0.05 | UMA <0.02, OrbMol <0.03, AIMNet2 >0.05 | 相比AceFF-1.0在带电分子上显著改善 |
| 分子动力学速度(3000原子水盒子,RTX4090) | 步/秒 | AceFF-2约50-75步/秒 | OrbMol约30, ANI-2x约20, MACE-MPA-0 <10 | 小体系(<300原子)可达>100步/秒,得益于CUDA图 |
| MLIP/MM混合动力学(蛋白-配体复合物,RTX4090) | 速度 (ns/day) | 36.7(2fs步长)/ 72.6(1fs步长) | ANI-2x 59.1, AceFF-1.0 63.8, MACE-MPA-0 4.2 | 比MACE-MPA-0快8-17倍,速度接近ANI-2x |
| Platinum Diverse构象最小化(2859结构) | 平均RMSD (A) / %<1A | 0.552 A / 86.29% | AIMNet2 0.508/89.75%, OrbMol 0.534/86.92% | 与OrbMol无统计显著差异(KS检验p=0.064) |
局限与改进
论文承认了多个局限性。首先,AceFF-2在某些极端情况下会失败:在Platinum Diverse基准中,2个分子(电荷分别为-8和-3,含多个磷酸基团)的最小化产生了非物理的'爆炸'结构。这是因为训练数据中分子电荷范围仅为[-2, 2],这些分子远超出模型的知识域。其次,推理速度方面,AceFF-2的库仑项实现了$O(N^2)$缩放,这意味着对于大体系(>3000原子)可能会比线性缩放的模型更慢。第三,当前模型无法用于完整的生物分子模拟(>10,000原子),只能用于MLIP/MM混合模拟中的配体区域。第四,在Schrodinger配体测试中,能量误差未能降到0.05 eV以下,作者承认这是因为测试分子(27-74个原子)大于训练数据中的最大分子(30个原子)。第五,OrbMol和UMA的优异表现部分归因于它们在更大的OMol25数据集(1亿构象,比AceFF数据集大约10倍)上训练,可能包含测试分子或非常相似的分子。此外,作者指出他们未测试直接力预测MLIP(力在前向传播中输出而非通过反向传播),因为这类模型的非保守性会给稳定的MD模拟带来额外复杂性。最后,与OpenMM-ML的兼容性受限——OrbMol不兼容TorchScript,AIMNet2尚未在OpenMM-ML中实现。
独立分析的弱点
尽管AceFF-2表现出色,但仍存在几个值得关注的弱点。第一,电荷范围限制在[-2, 2],这在实际药物发现中是一个实际问题——许多药物分子含有多个磷酸基团、磺酸基团等,总电荷可能远超-2。改进方向包括扩展训练数据的电荷范围,或者在模型中加入对超出训练域的电荷值的鲁棒性机制。第二,库仑项的$O(N^2)$实现限制了模型在大体系中的应用。可以考虑使用Ewald求和、粒子网格Ewald(PME)或快速多极子方法(FMM)将长程静电相互作用的计算复杂度降至$O(N\log N)$或$O(N)$。第三,训练数据中分子最大仅30个原子,这限制了模型对更大药物分子的外推能力。虽然测试显示力误差大部分可接受,但能量误差明显偏高。可以通过增加更大分子的训练数据或采用分层训练策略来改善。第四,模型仅在真空和隐式溶剂条件下训练,未考虑显式溶剂效应,这可能影响模拟的物理真实性。第五,CUDA图兼容性虽然带来速度优势,但限制了模型架构的灵活性——不是所有操作都与CUDA图兼容。
未来方向
作者在论文中提出了几个明确的未来方向。首先,他们提到可以将TensorNet2的某个电荷通道在QM级别的部分电荷(如MBIS电荷)上进行训练,以支持更精细的静电相互作用建模。其次,他们指出OMol25数据集包含Mulliken和Lowdin电荷,不同的电荷通道可以同时在多种QM衍生的部分电荷上训练。更广泛地说,基于AceFF-2的成果可以延伸以下方向:(1)将模型扩展到更大的分子体系,可能采用分层或自适应精度策略;(2)开发兼容OpenMM-ML的实现,以便更广泛地用于生物分子模拟;(3)结合自由能微扰(FEP/RBFE)方法进行大规模药物发现基准测试;(4)探索将AceFF-2与显式溶剂力场结合的更精确的混合模拟方案;(5)开发支持CUDA图的直接力预测版本以进一步提高速度;(6)将TensorNet2的电荷平衡思想扩展到其他等变架构(如MACE、NequIP)。
复现评估
在复现性方面,本文提供了良好的开源支持。TensorNet2架构在MIT许可证下开源,代码在https://github.com/torchmd/torchmd-net。AceFF模型权重可在HuggingFace下载(https://huggingface.co/Acellera/AceFF-2.0)。作者提供了IPython笔记本教程(可在Google Colab运行)和Python包,支持ASE计算器接口和OpenMM-ML集成,代码在https://github.com/Acellera/aceff_examples。用于AIMNet2 CUDA图兼容性的修改也以PR形式提交。然而,训练数据集是内部构建的(约200万分子、1200万构象),并未公开,这限制了完全复现训练过程的可能性。算力方面,模型在RTX4090上进行推理测试,训练需要类似或更强的GPU资源,但由于模型较小(685,413参数),训练成本相对可控。总体而言,推理和使用可以完全复现,但完整的训练流程复现需要自行生成或获取类似规模的DFT数据集。
论文图表